Dynamics of aptamers
Leveraging NMR data to characterize the conformational landscape of aptamers
Eric Largy 
ARNA, INSERM U1212, CNRS UMR 5320, Université de Bordeaux
UFR des Sciences Pharmaceutiques, Université de Bordeaux
May 17, 2026
Binding loops stemming from wishful thinking
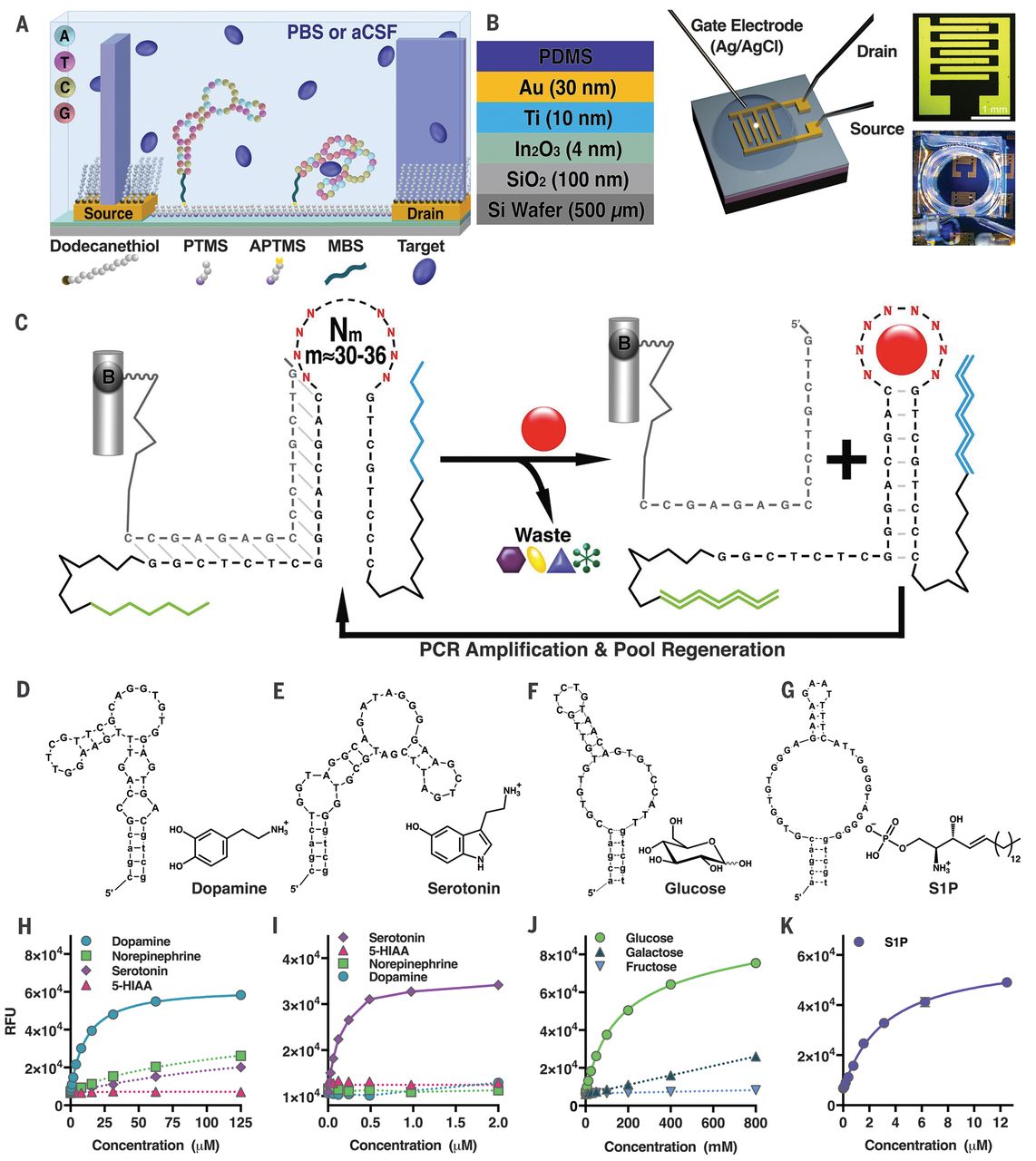
Binding loops stemming from wishful thinking
mFold produces this structure in 1 M NaCl solutions, at 37 °C
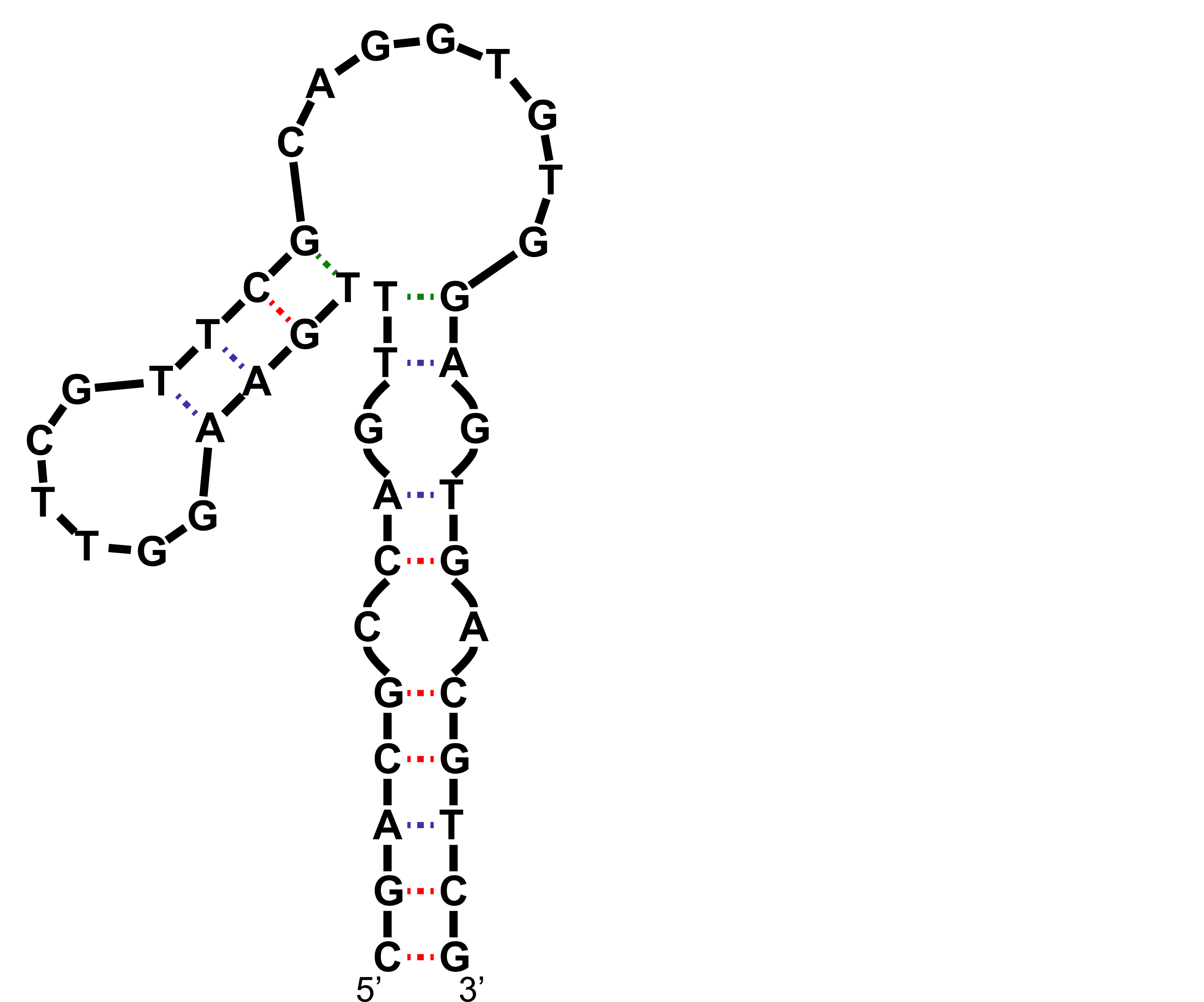
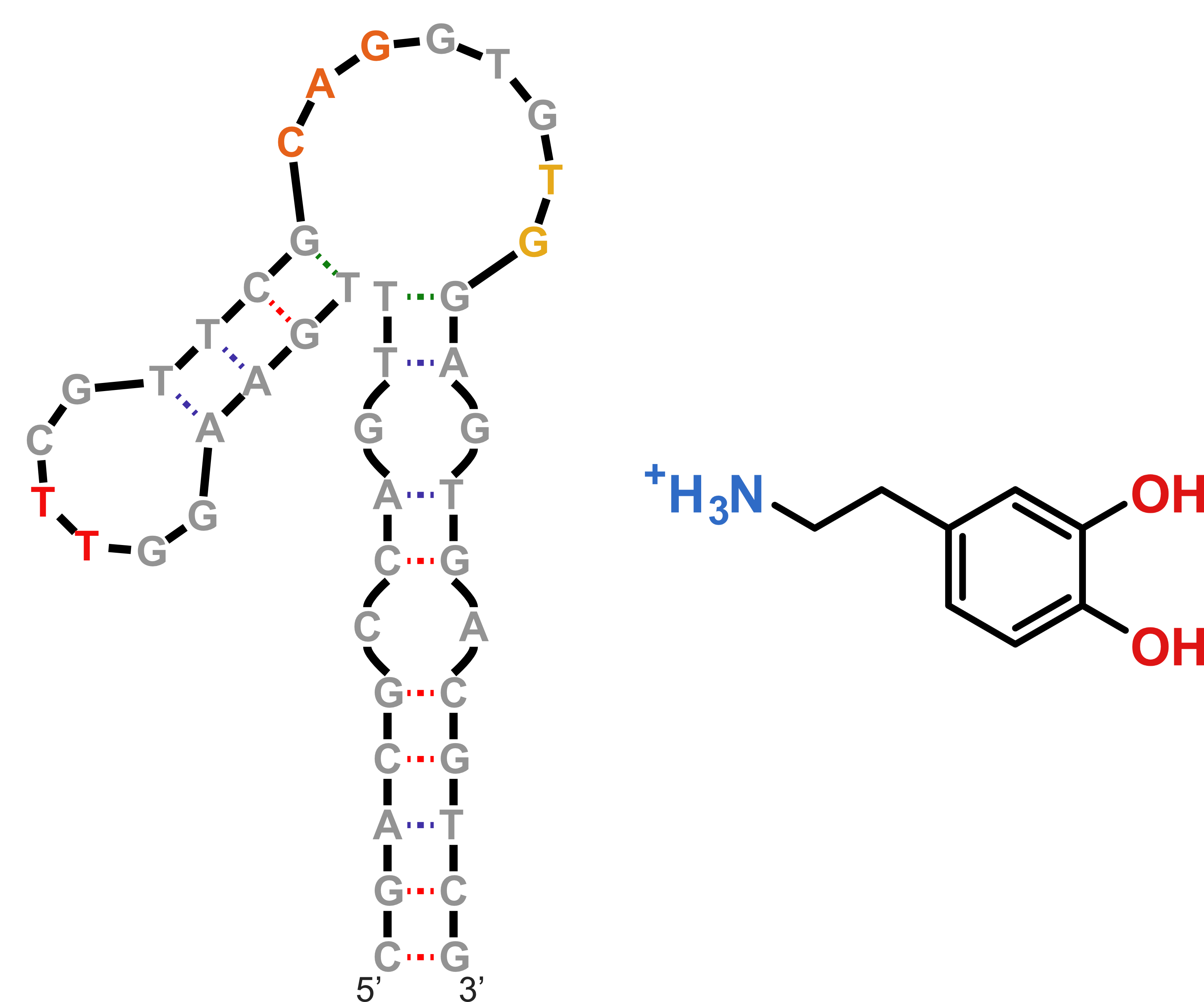
NMR provides a (non-canonical !) starting model
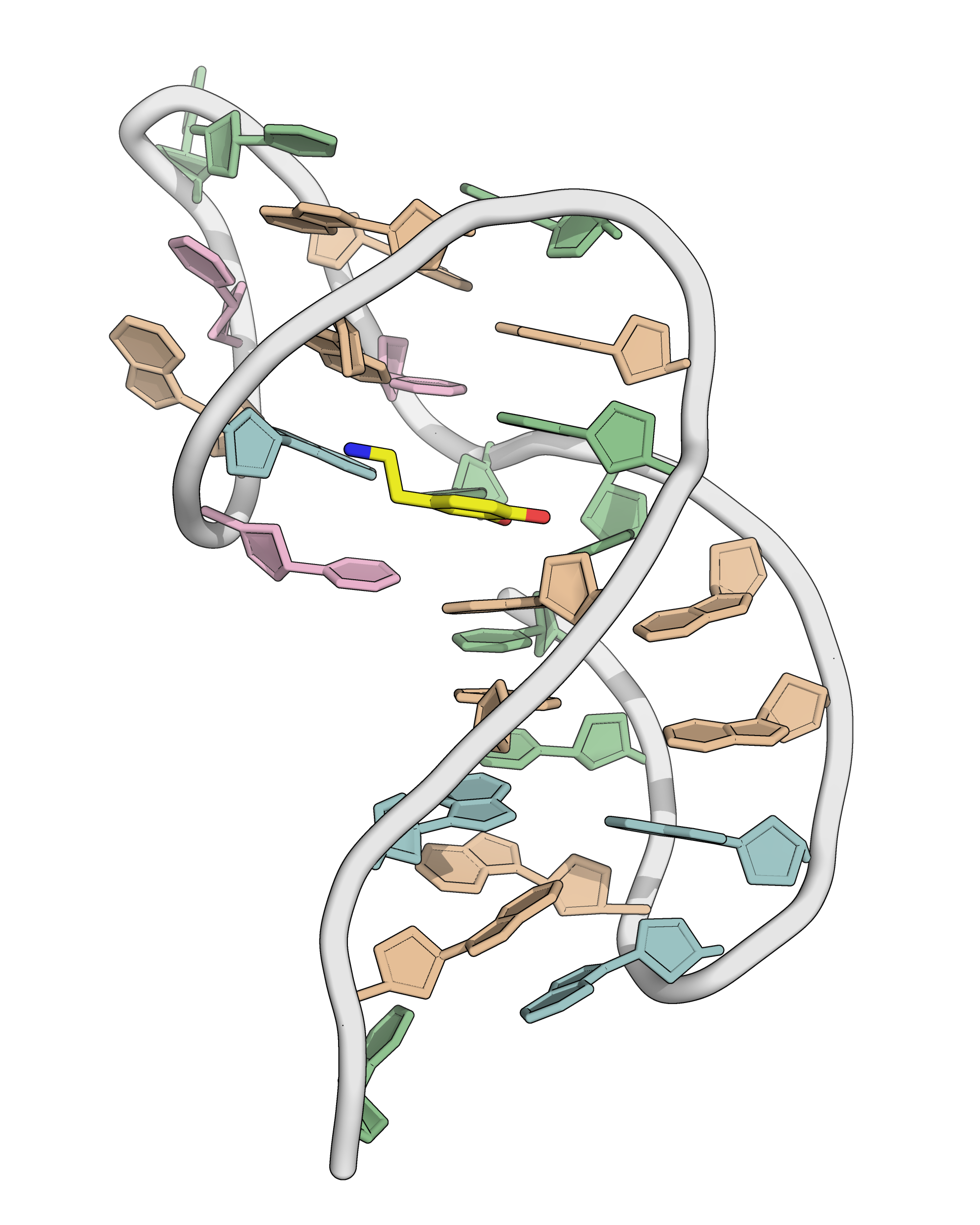
NMR provides an ensemble of models
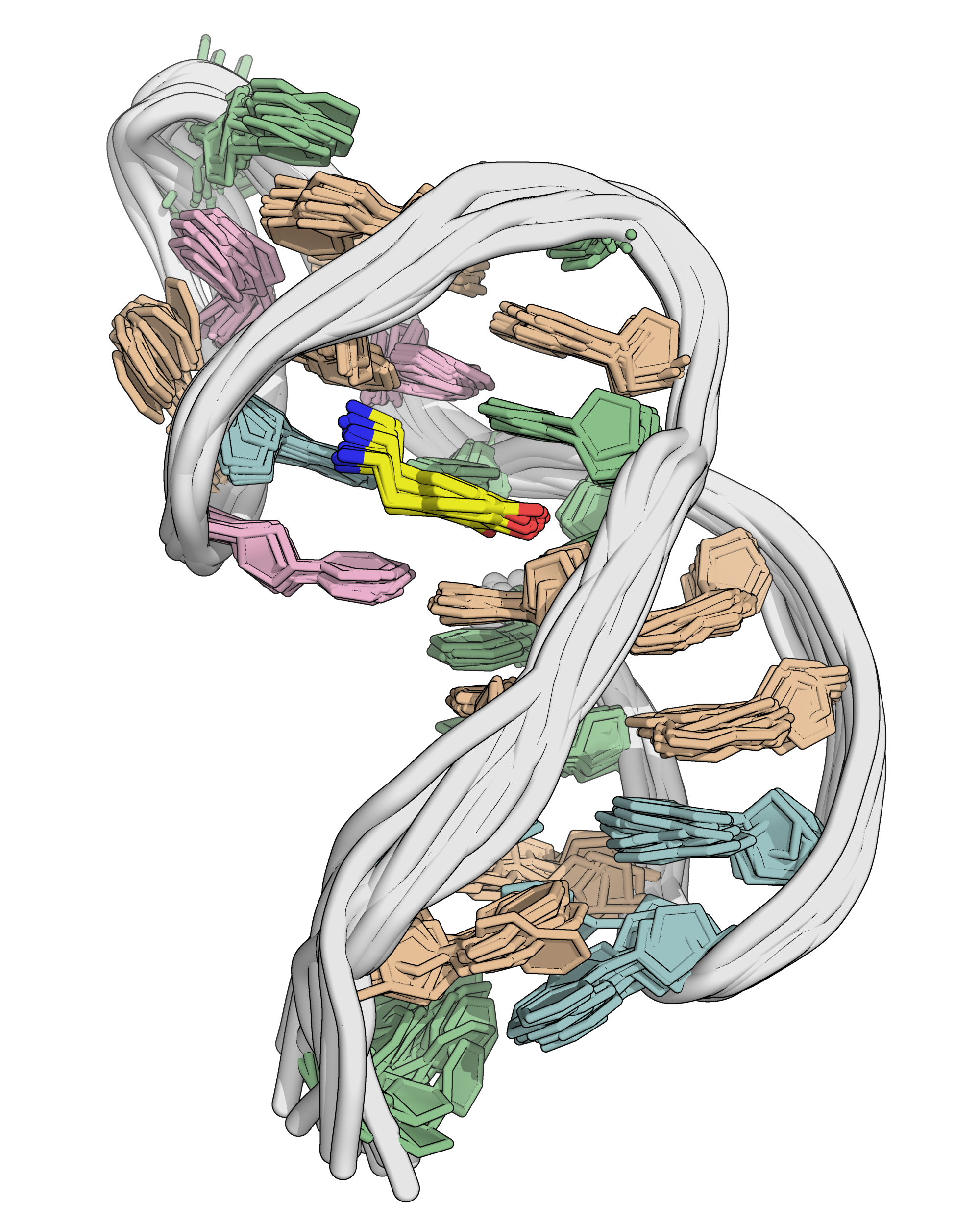
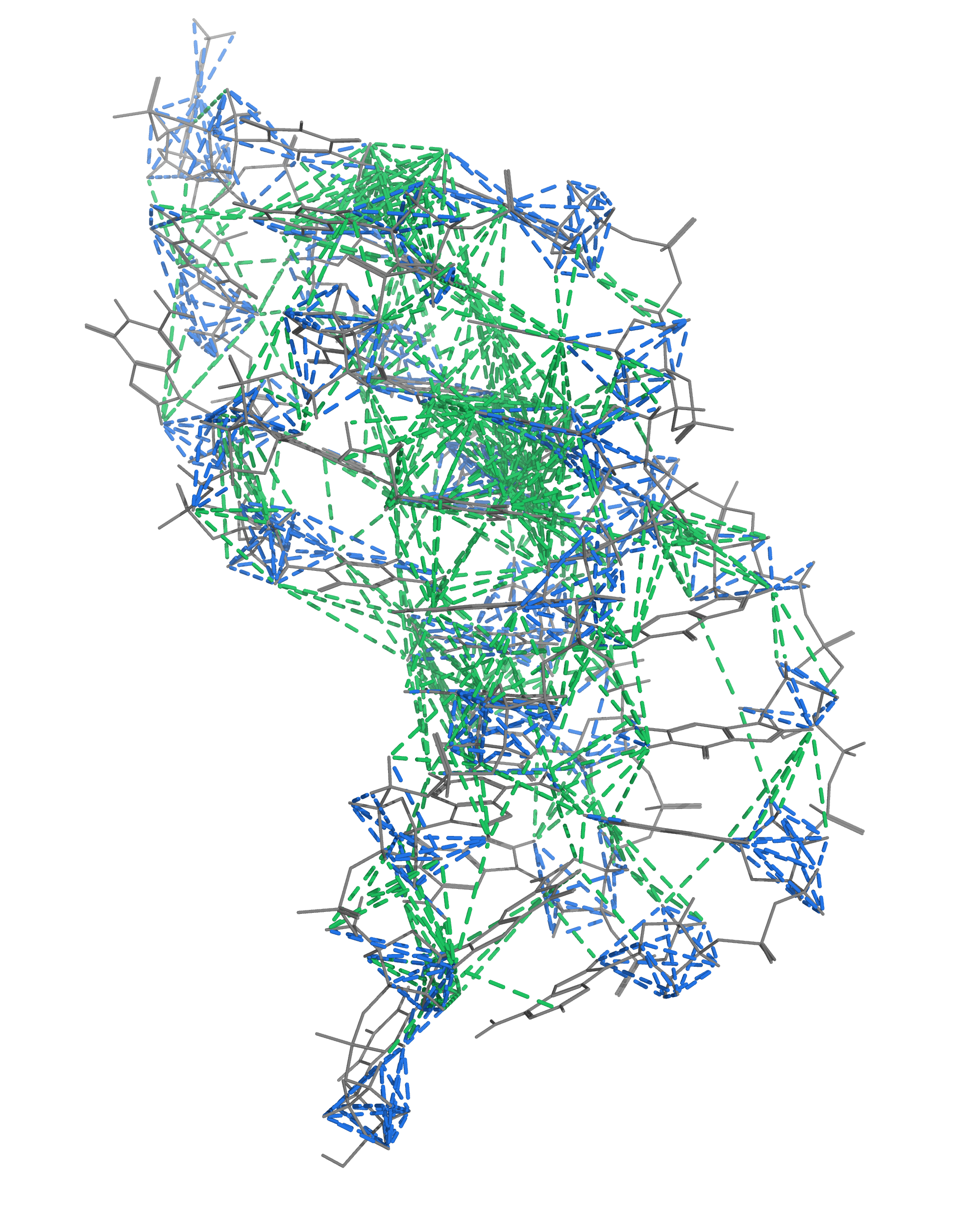
NMR provides many constraints
- DNA (806)
- intra-residue
- inter-residue
- Dopamine (67)
- intramolecular
- intermolecular
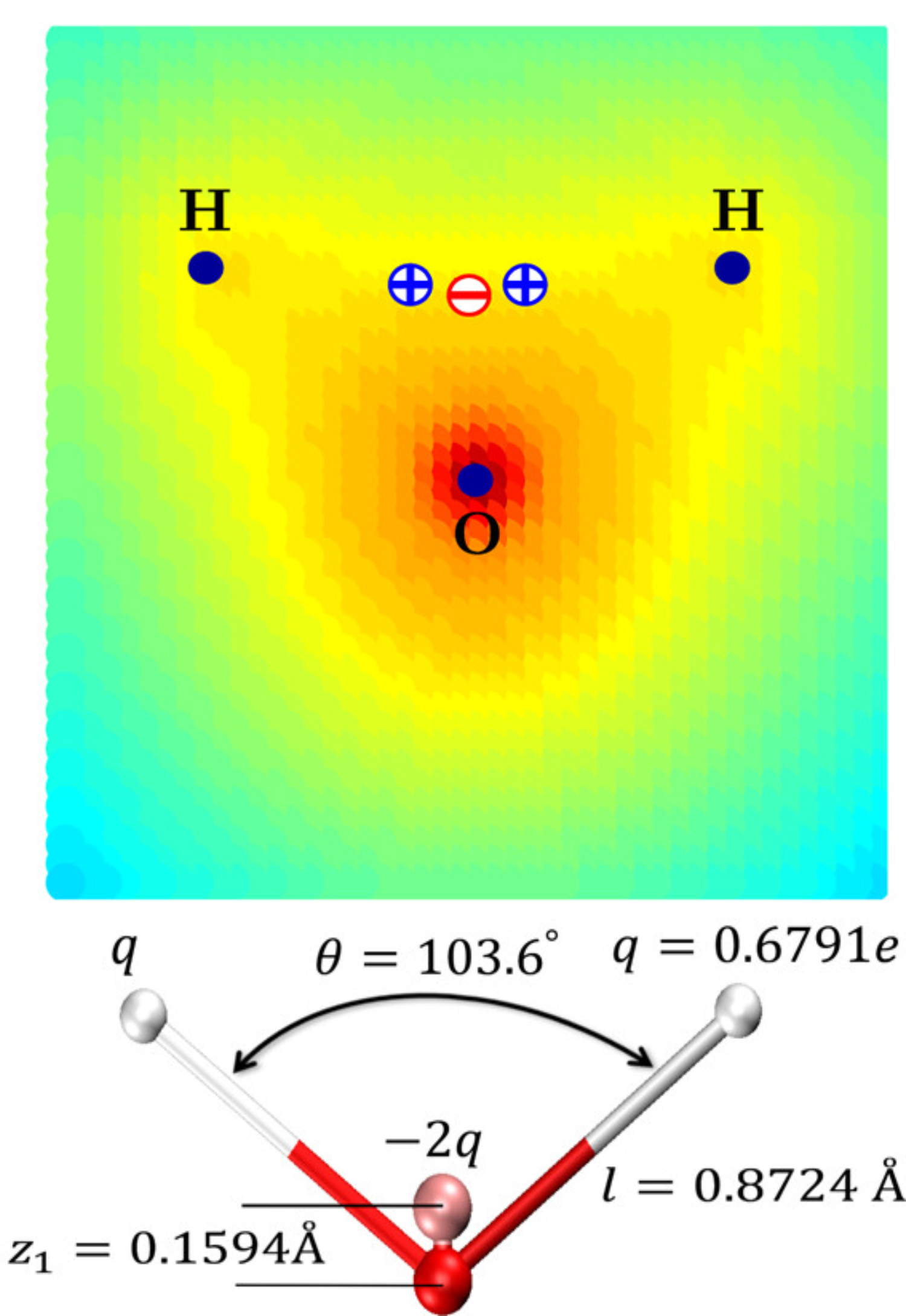
Solvation requires building a box with a model
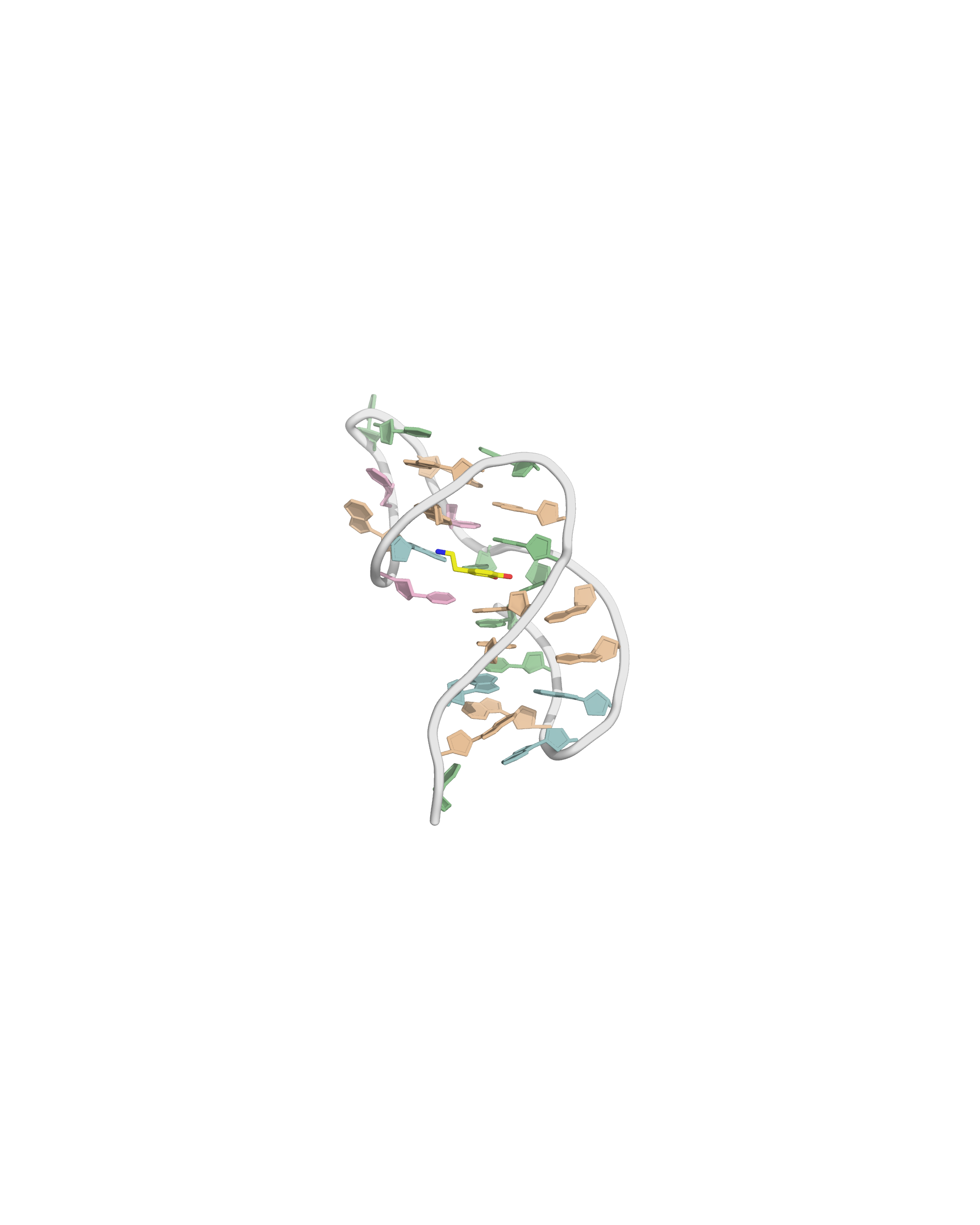
Solvation requires building a box with a model
- OPC water model
- Octahedral box with, 14 Å to surface

Na+ ions are added to neutralize the system
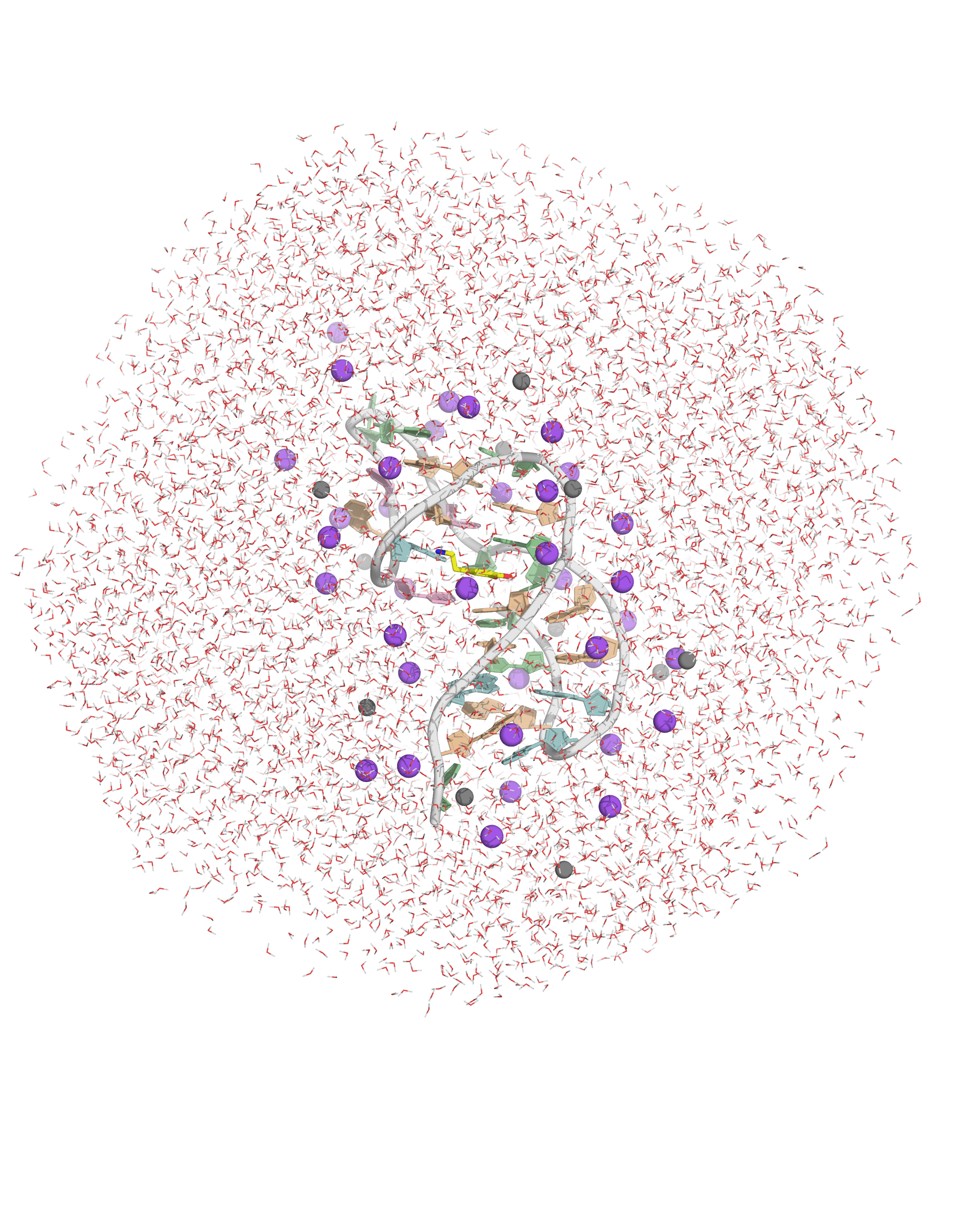
Na+ and Cl- are added to mimic experimental conditions
\(N_{\pm}=\color{#376bd3}{\nu_w}\color{#1a9c1a}{c_0}\color{#000000}e^{\mp \operatorname{arcsinh}\!\left(\frac{\color{#b92424}{Q}}{2\color{#376bd3}{\nu_w}\color{#1a9c1a}{c_0}}\right)}\)
- \(\color{#376bd3}{\nu_w}\): water volume of the box (~ 3x105 Å3)
- \(\color{#1a9c1a}{c_0}\): salt concentration (140 mM)
- \(\color{#b92424}{Q}\): charge (-25)
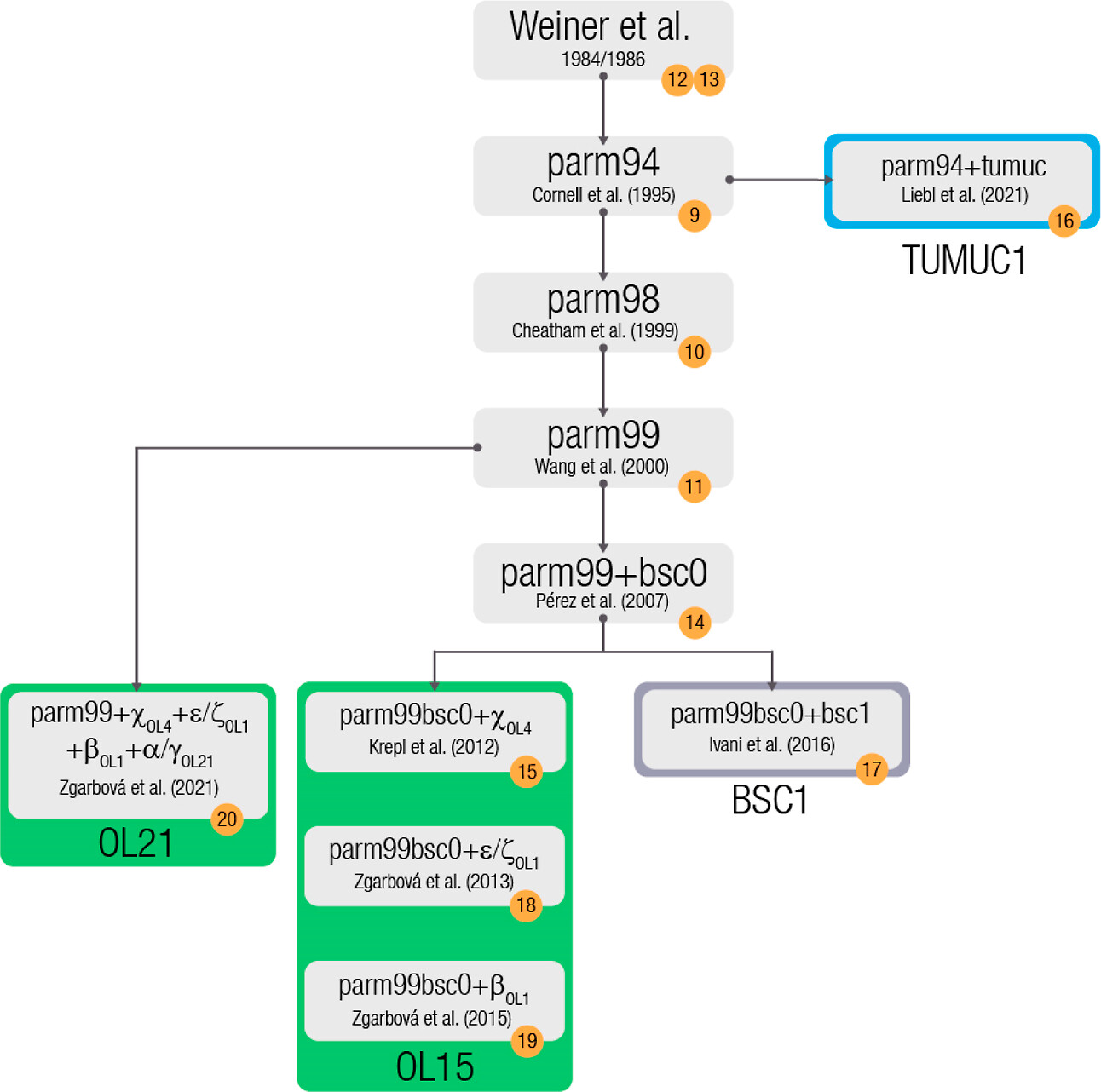
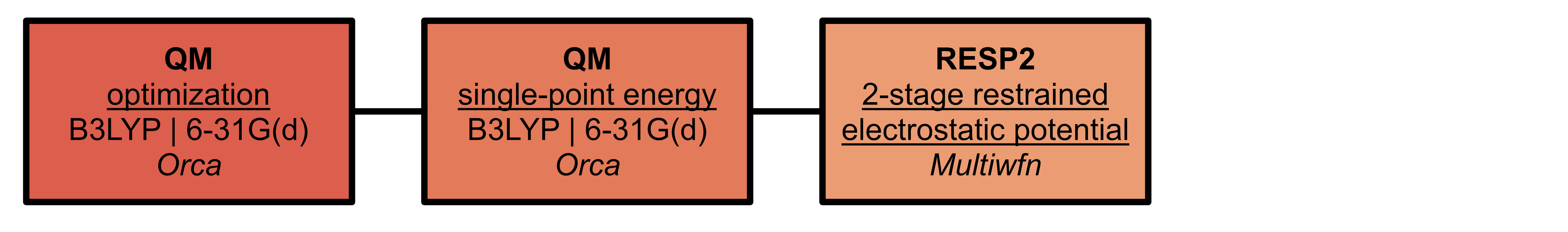
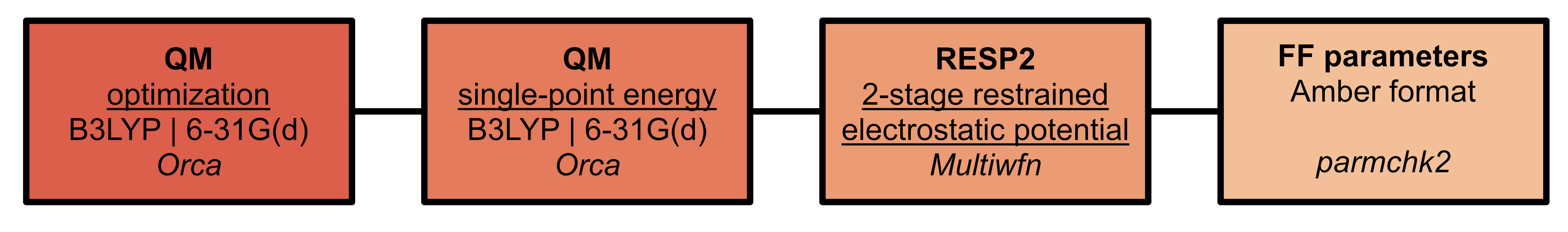
One or several force fields must be used for biomolecules
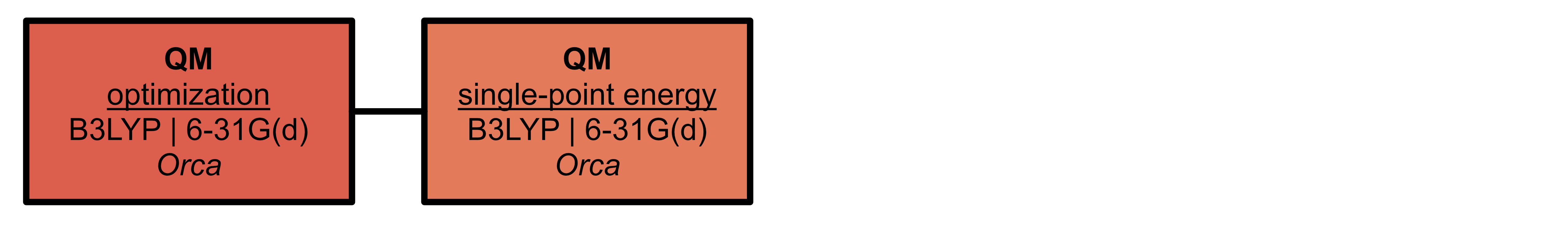
Where necessary, parameters for ligands must be generated

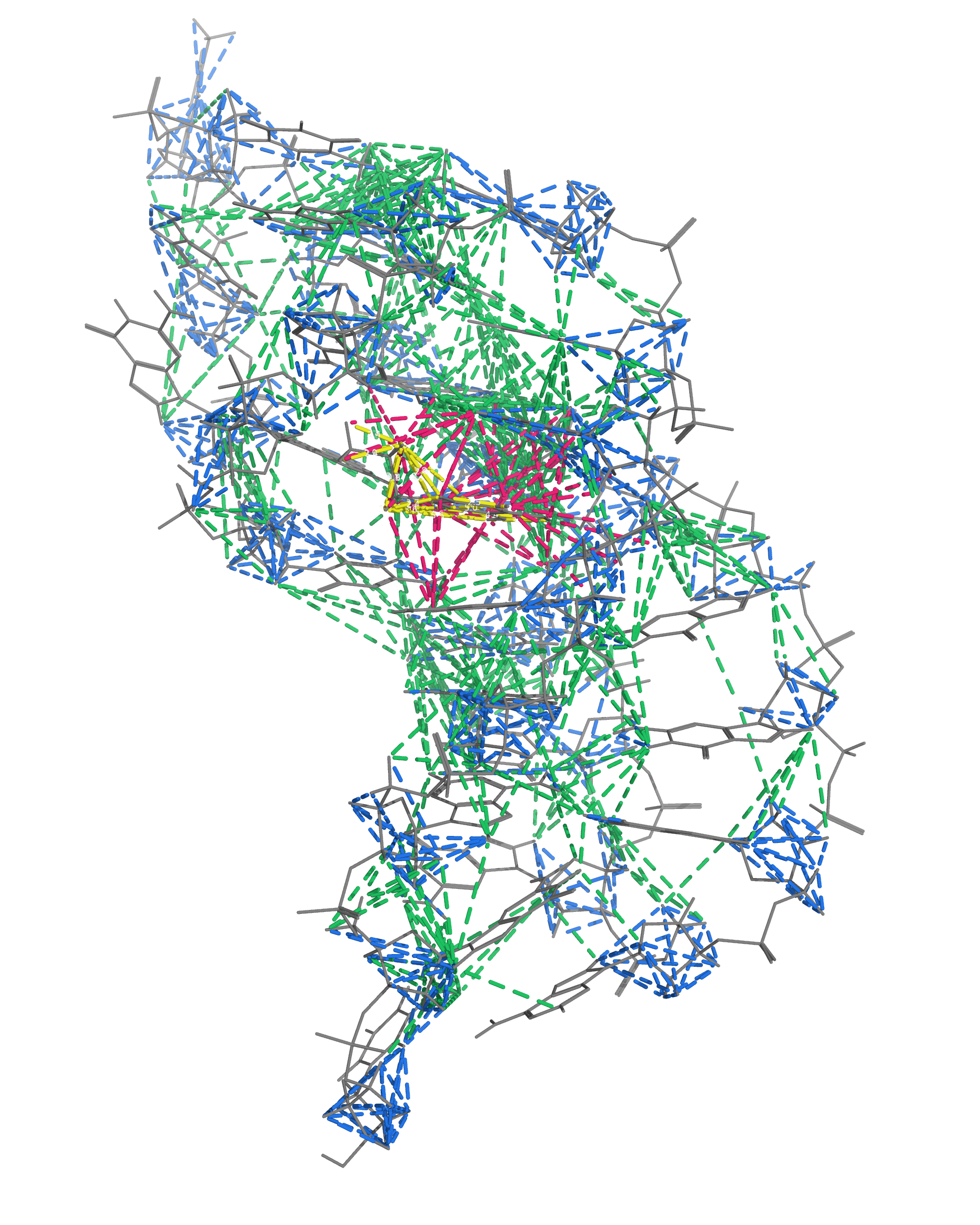
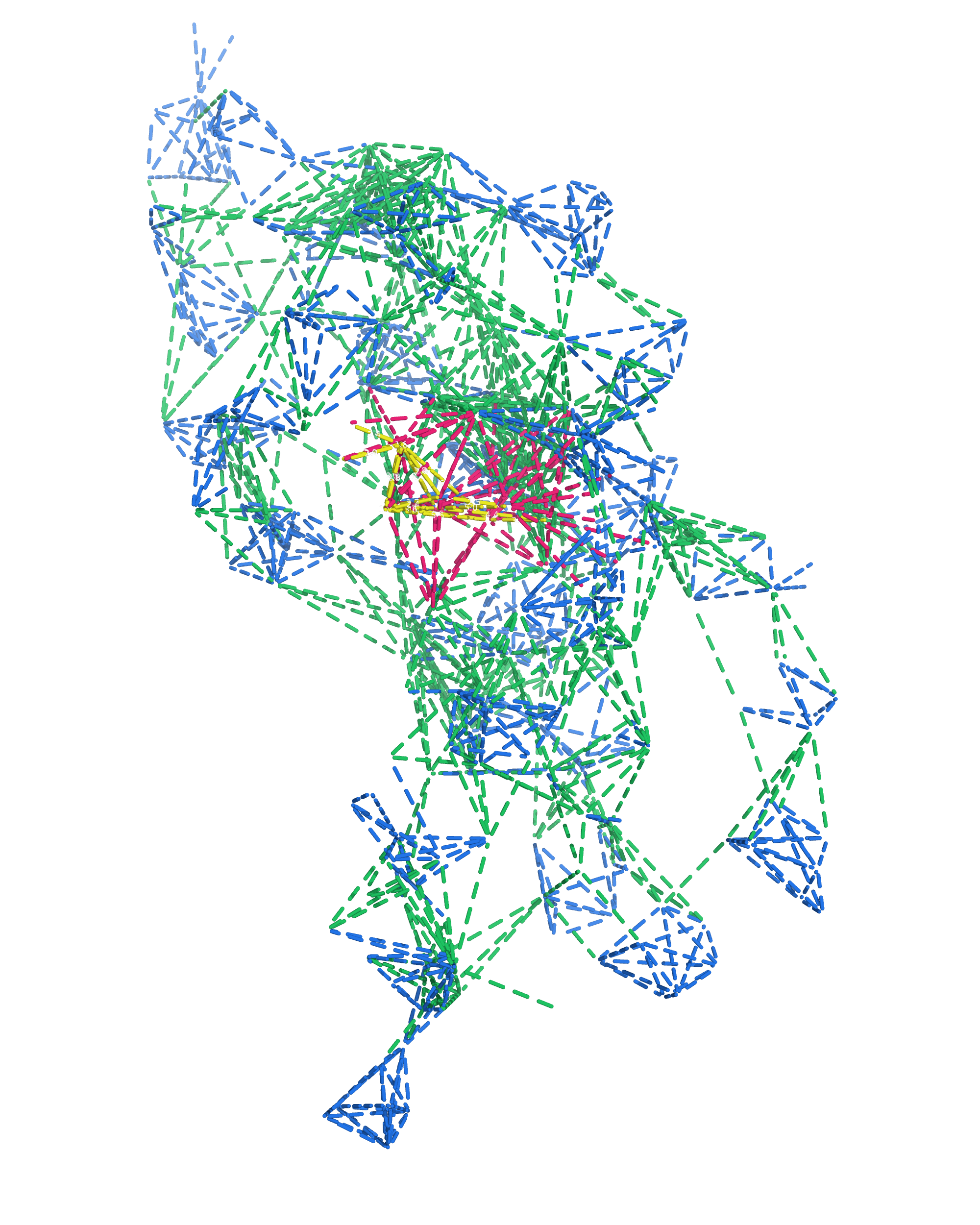
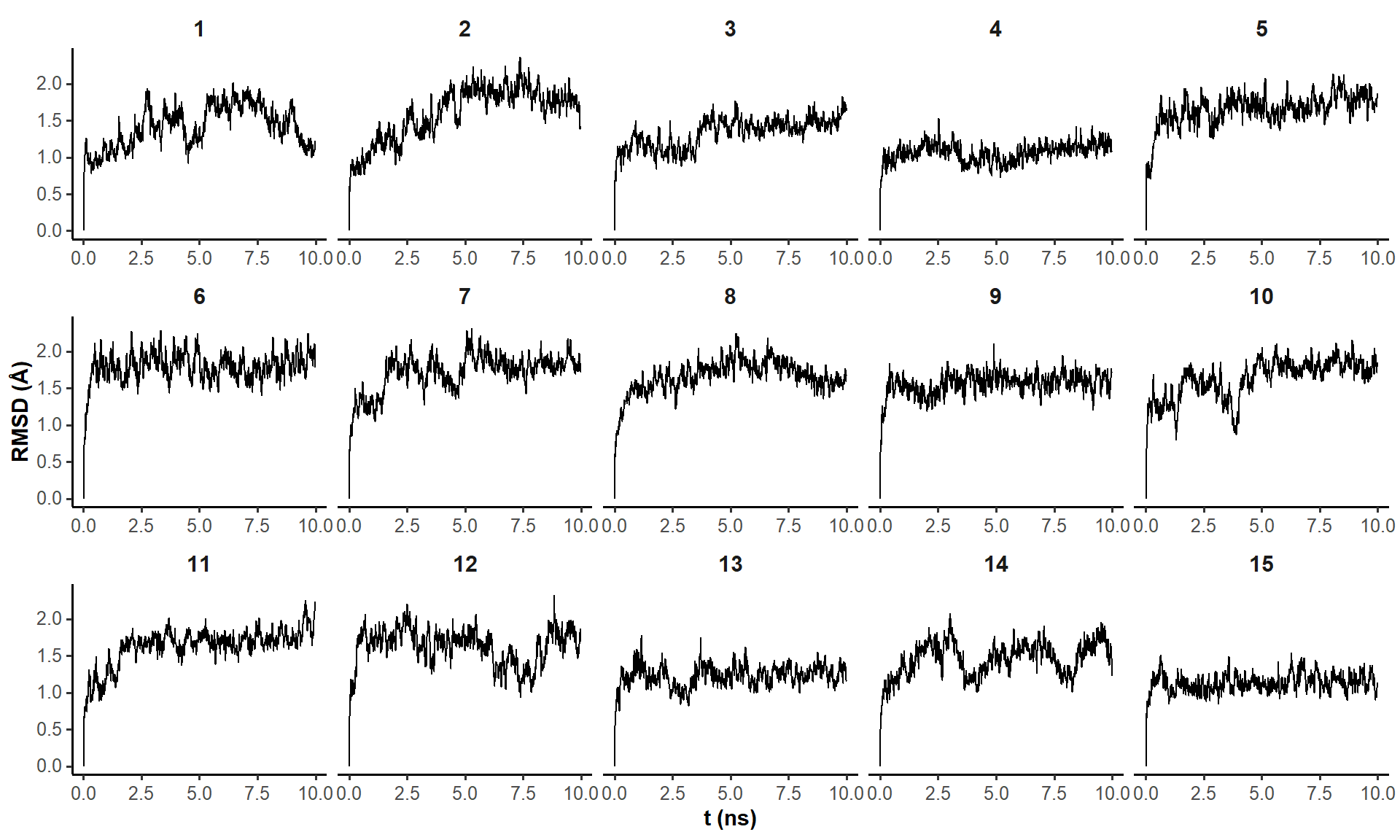
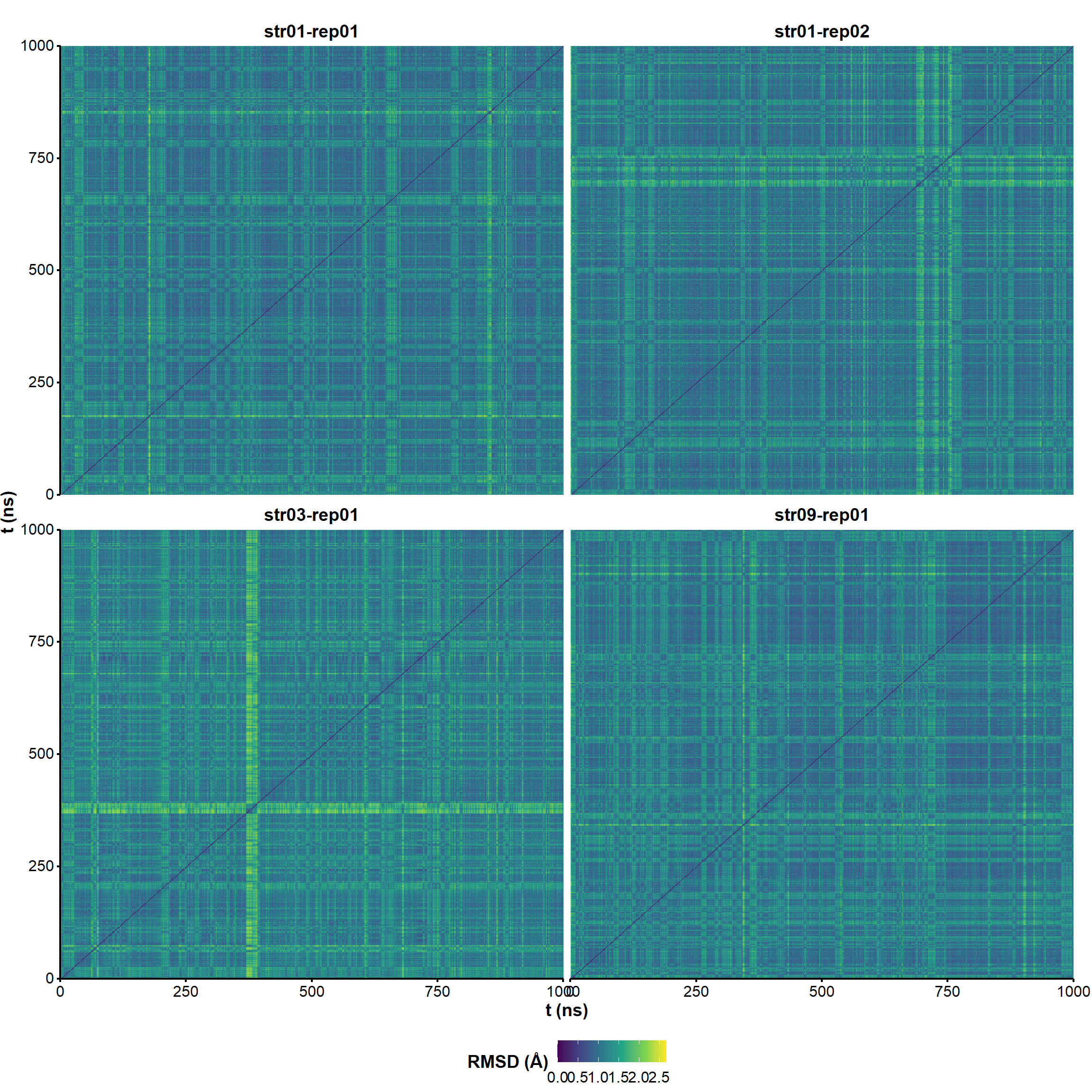
10-ns trajectories are stable RMSD-wise
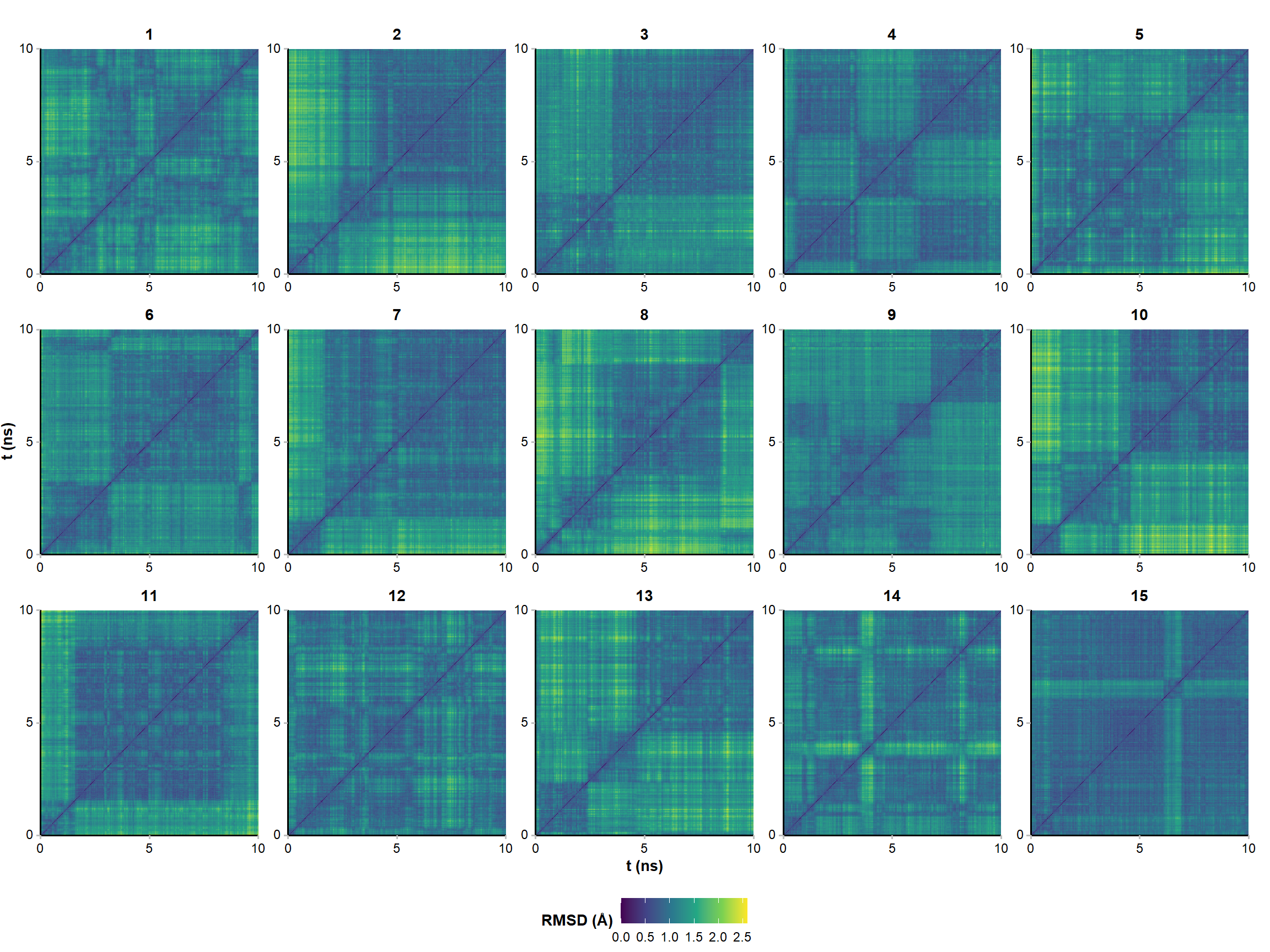
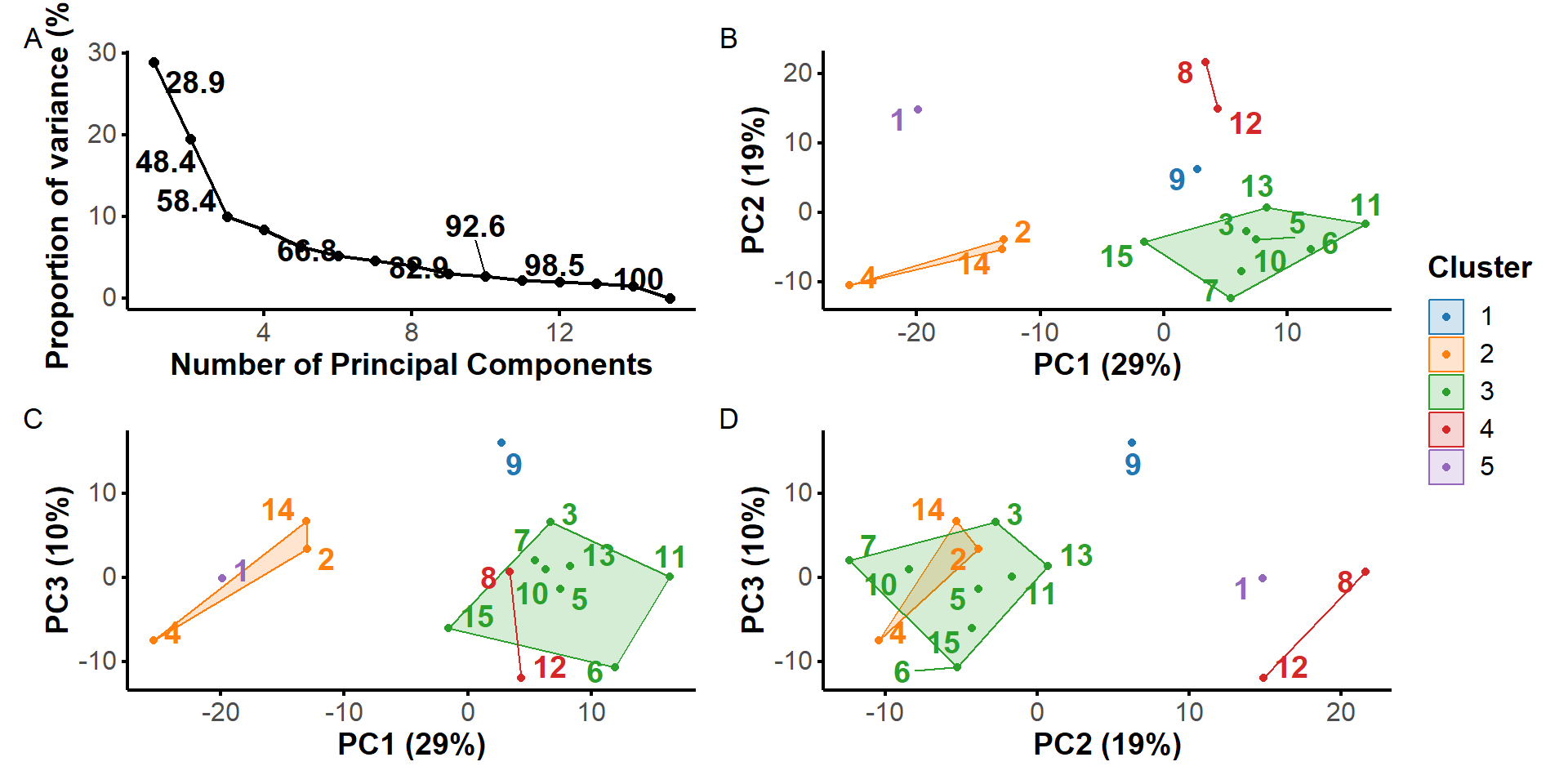
Pairwise RMSD analysis is more informative
The end point of 15 runs are minimized
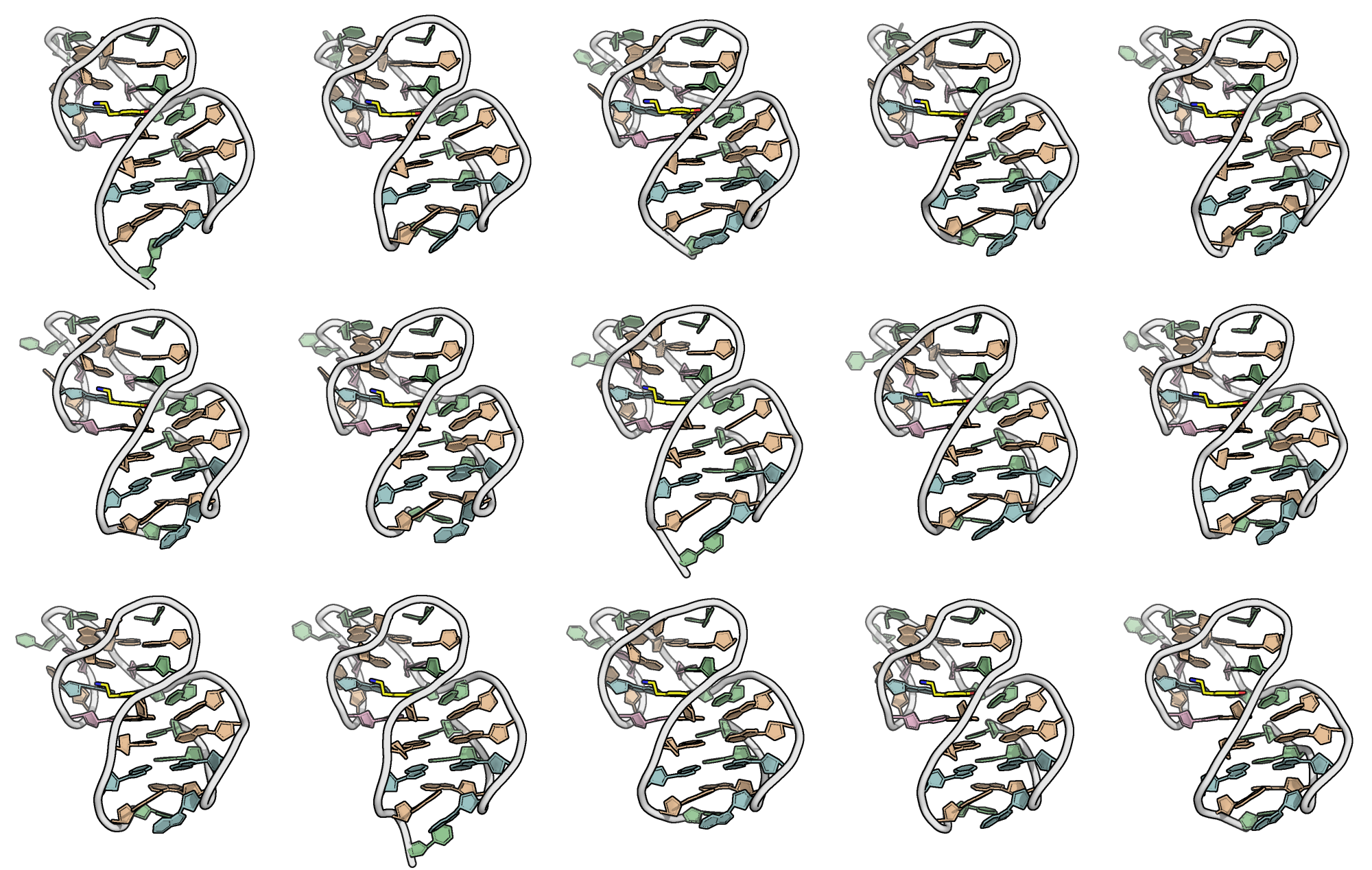
Superimposition reveals a well-defined binding pocket
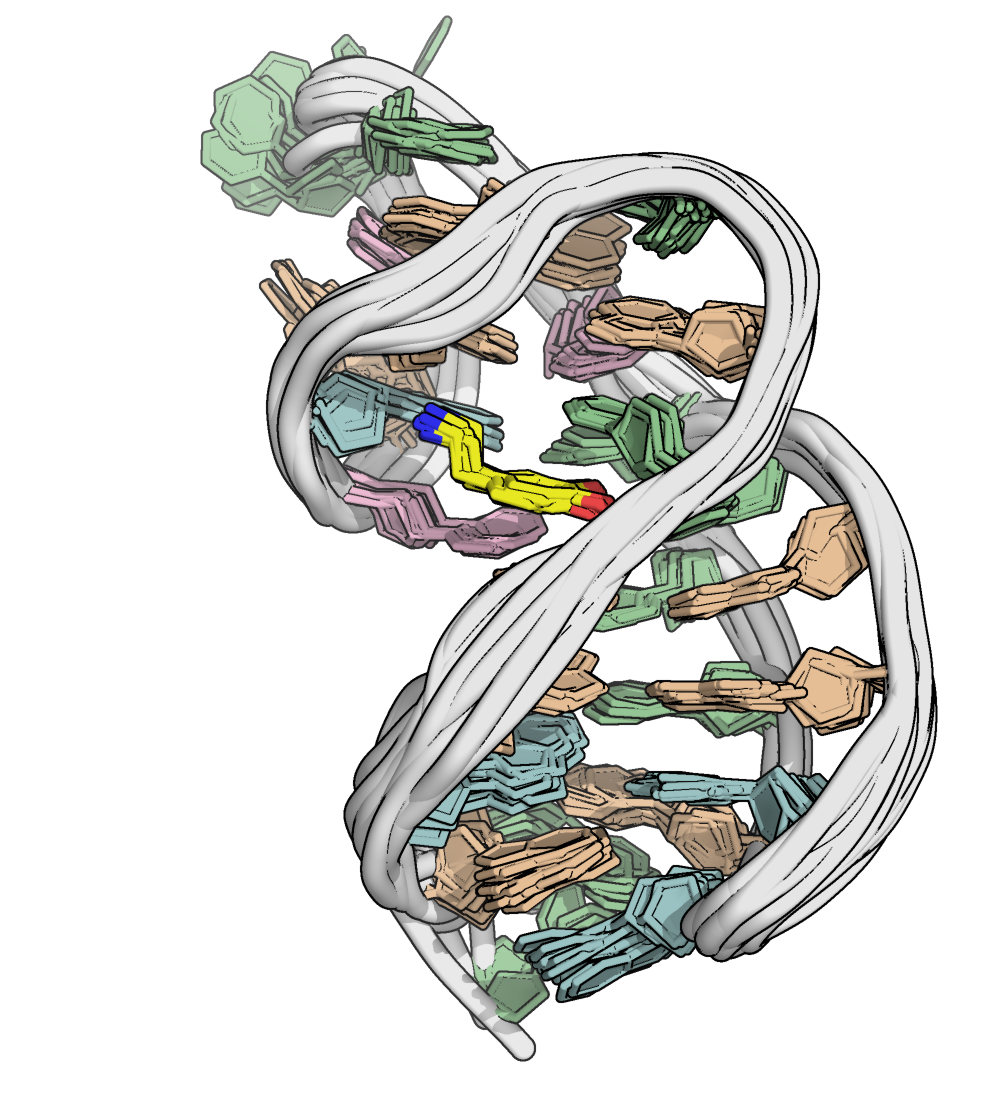
NMR restraints are satisfied
Replication is necessary (at least to publish!)
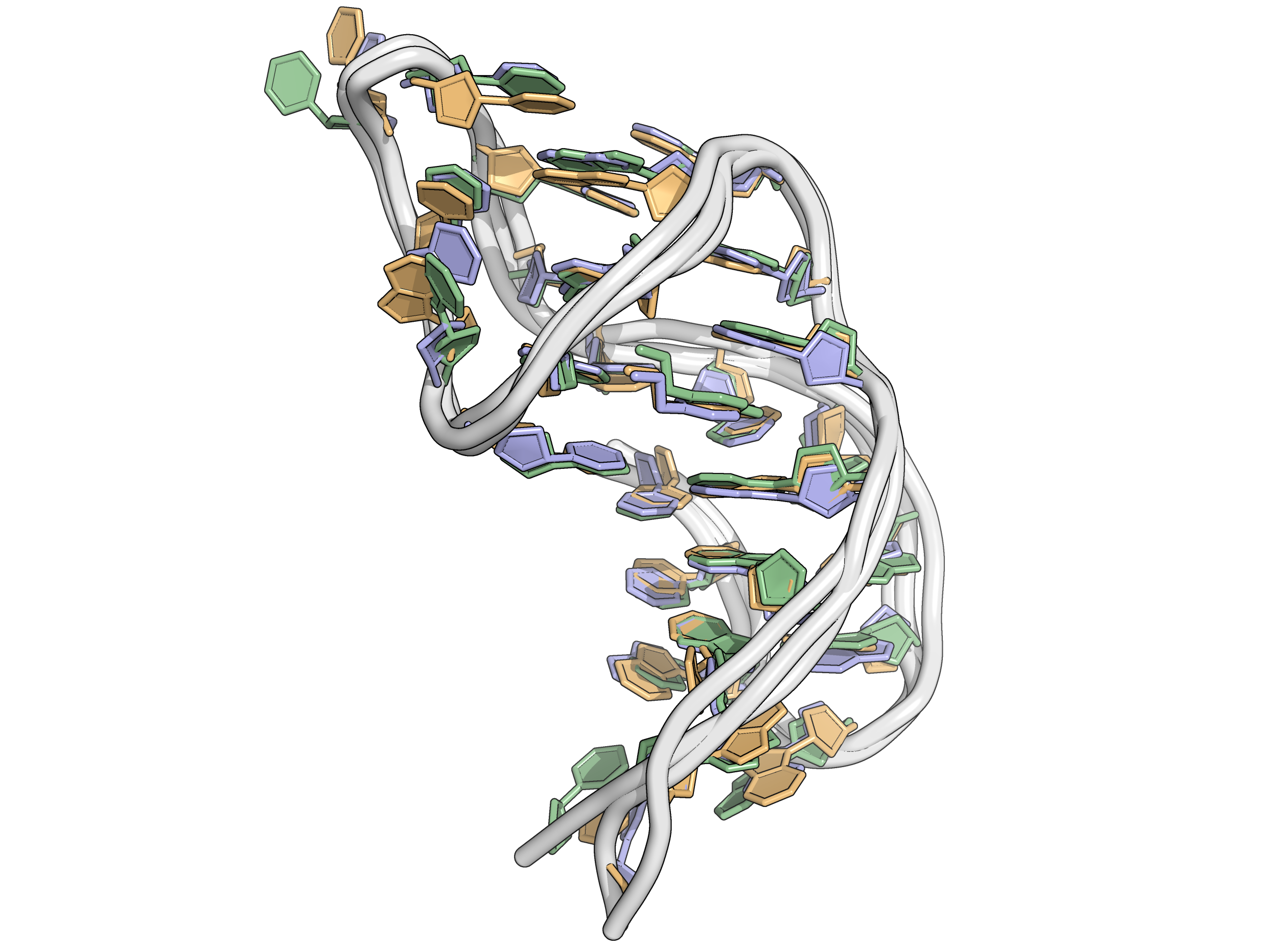
Longer simulations are stable and reproducible
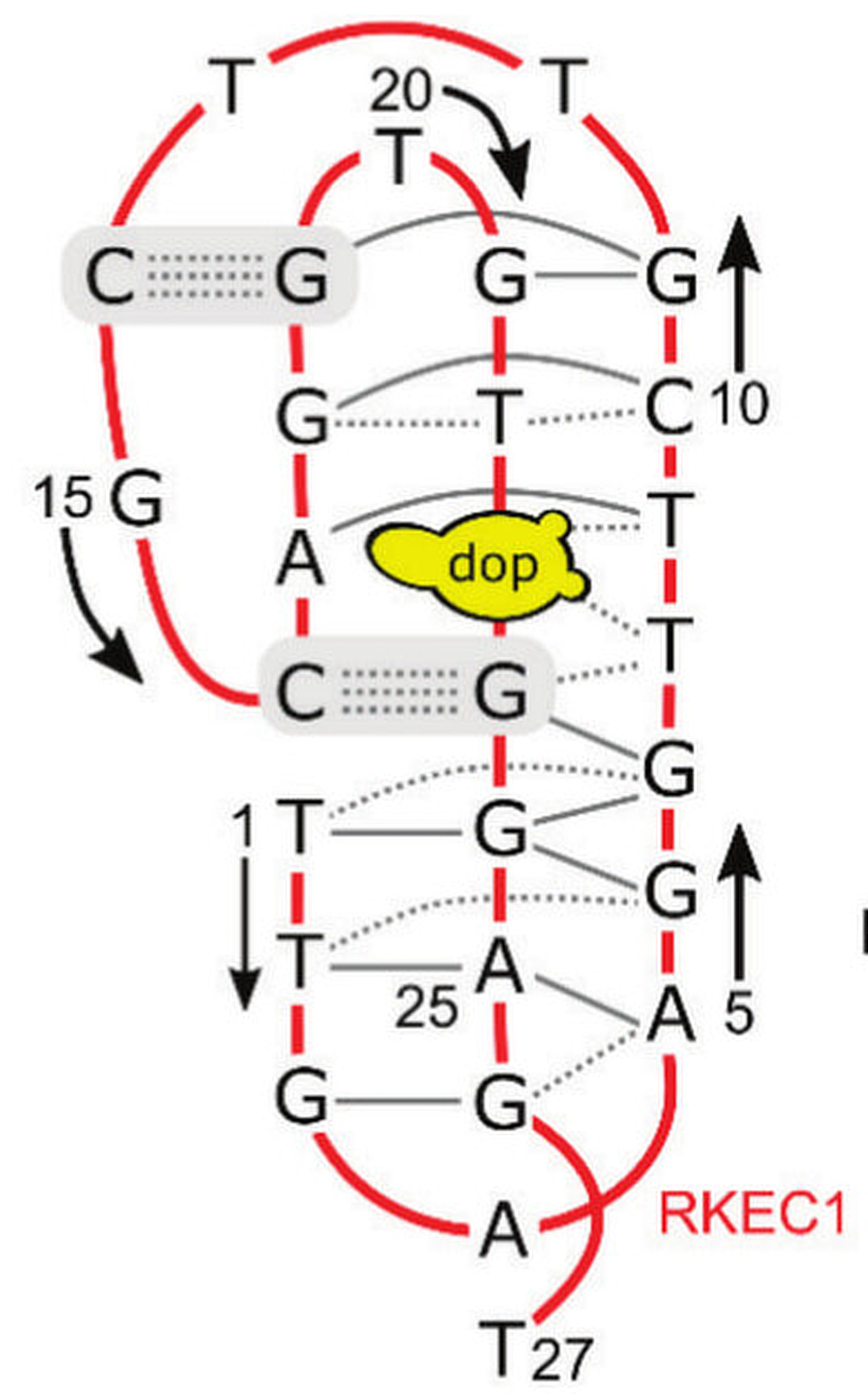
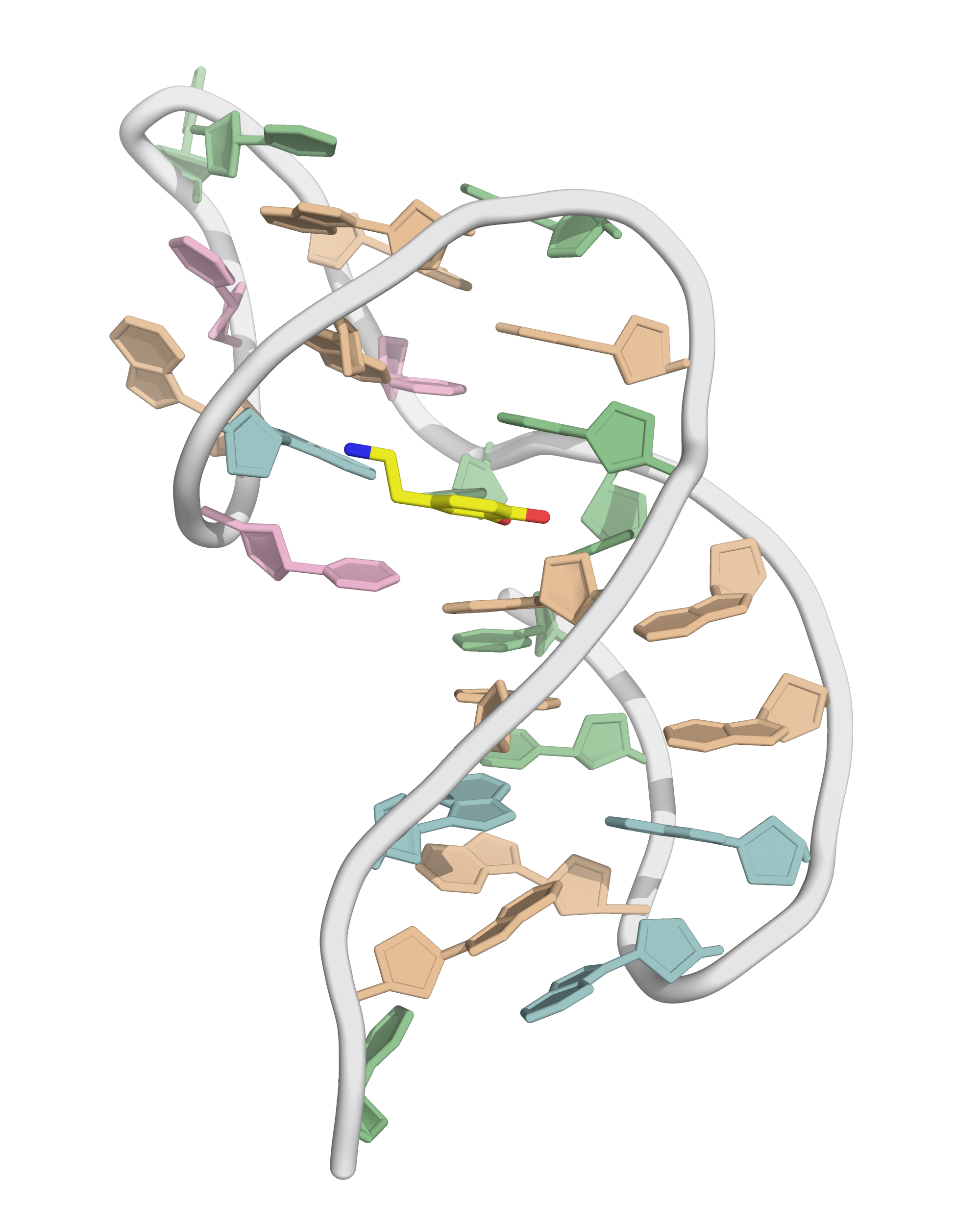
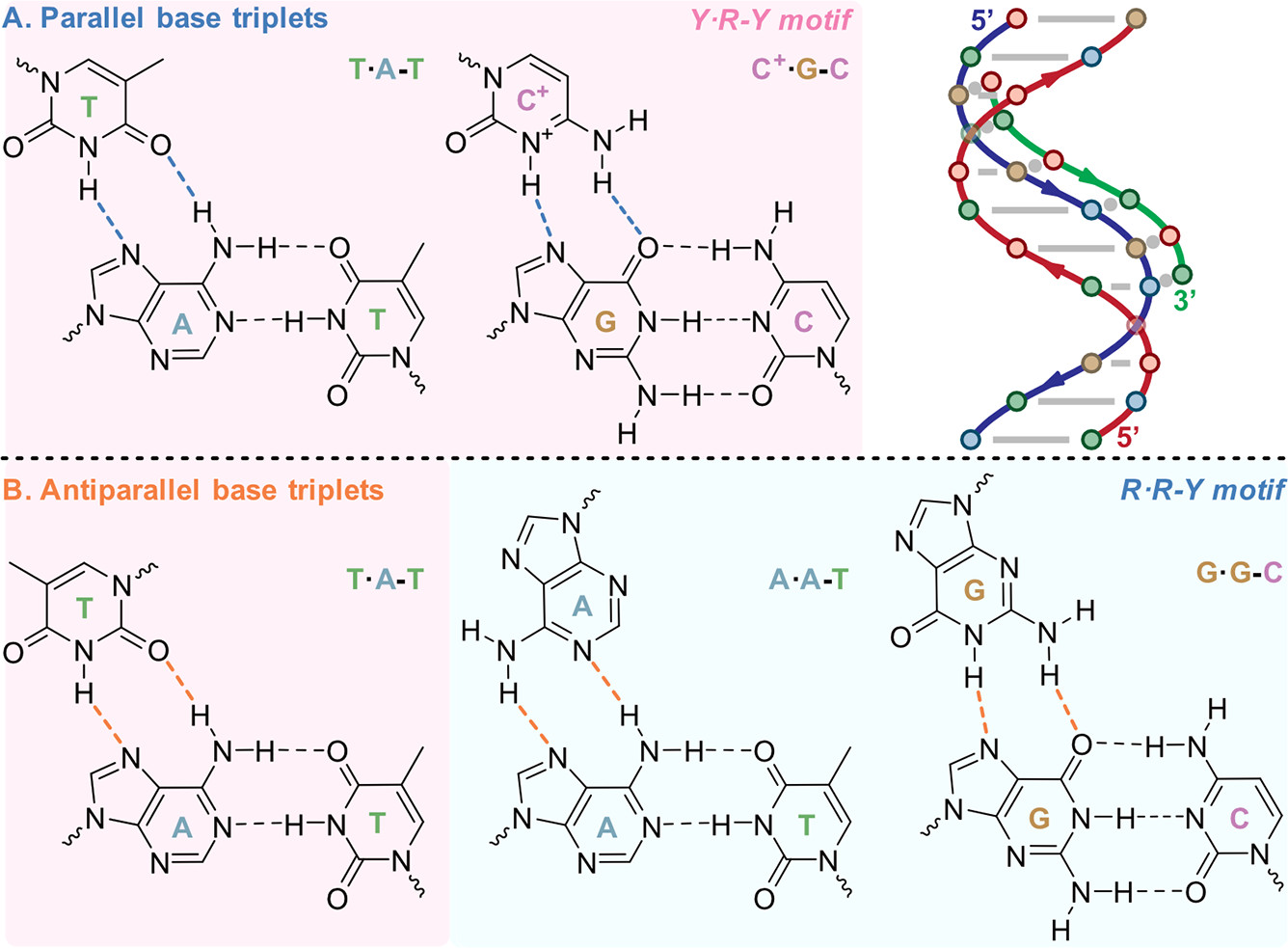
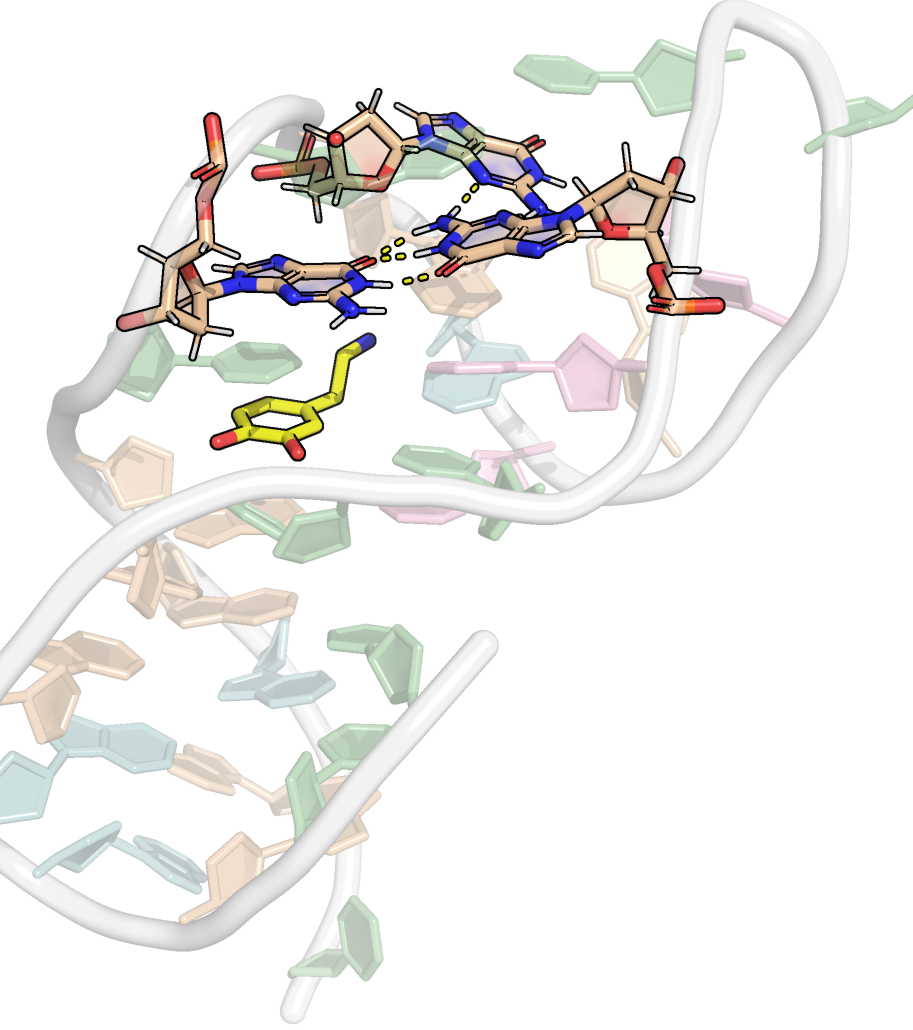
Non-WC base pairs and triplets
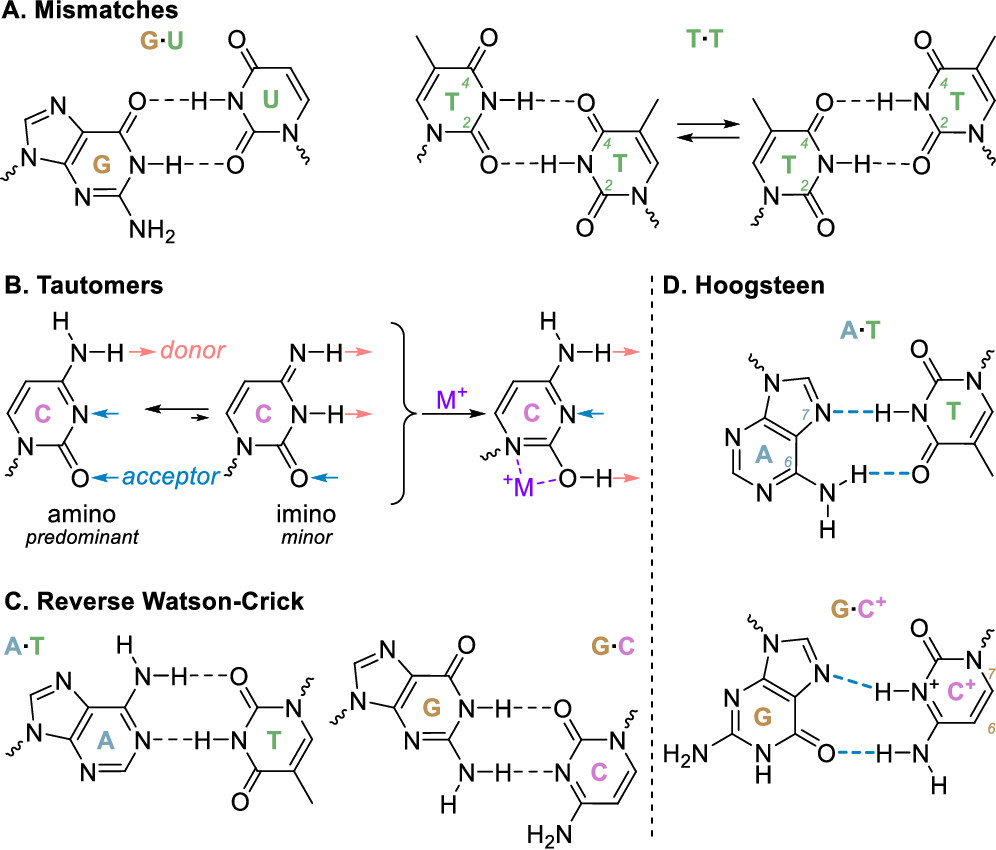

Only two WC base pairs are present
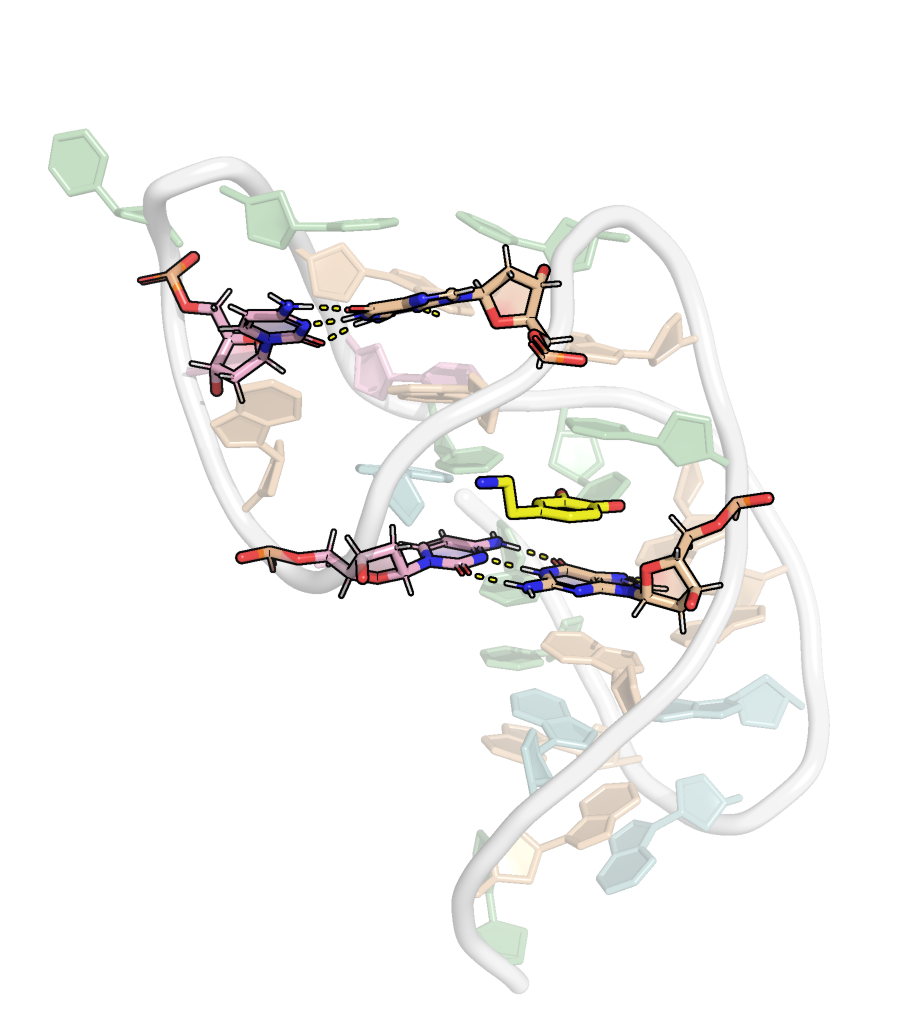
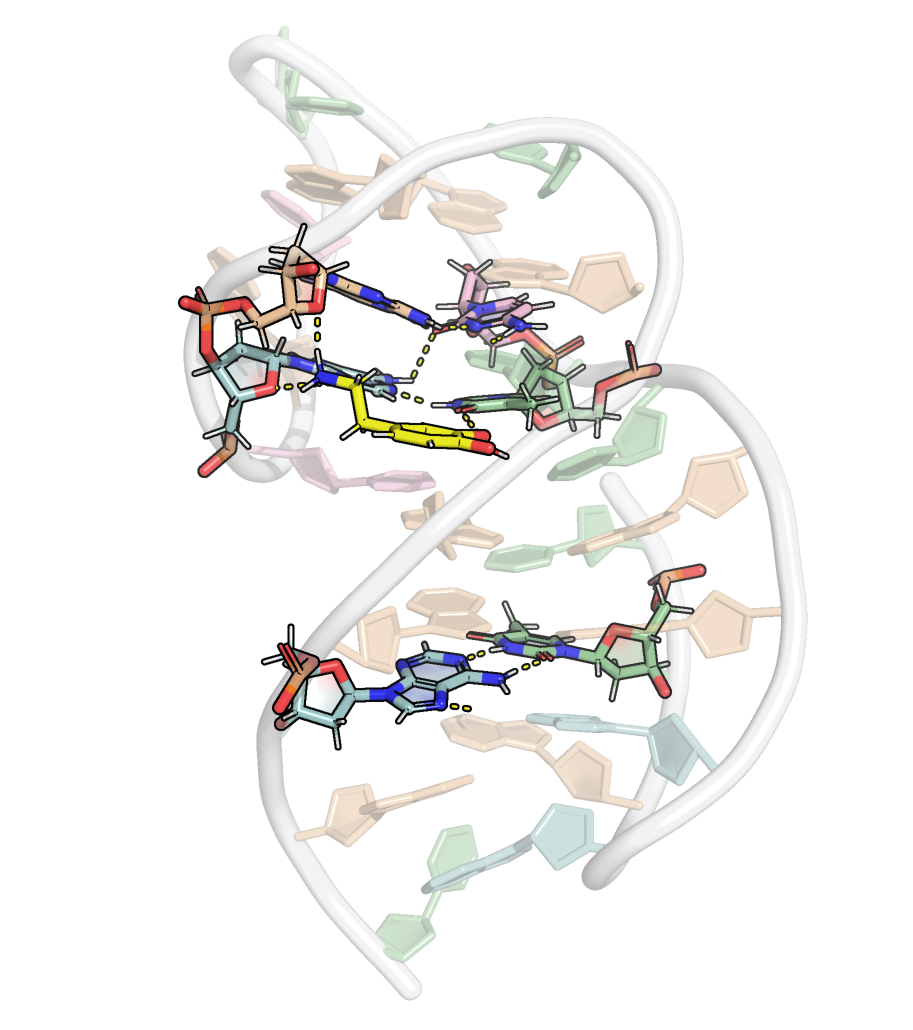
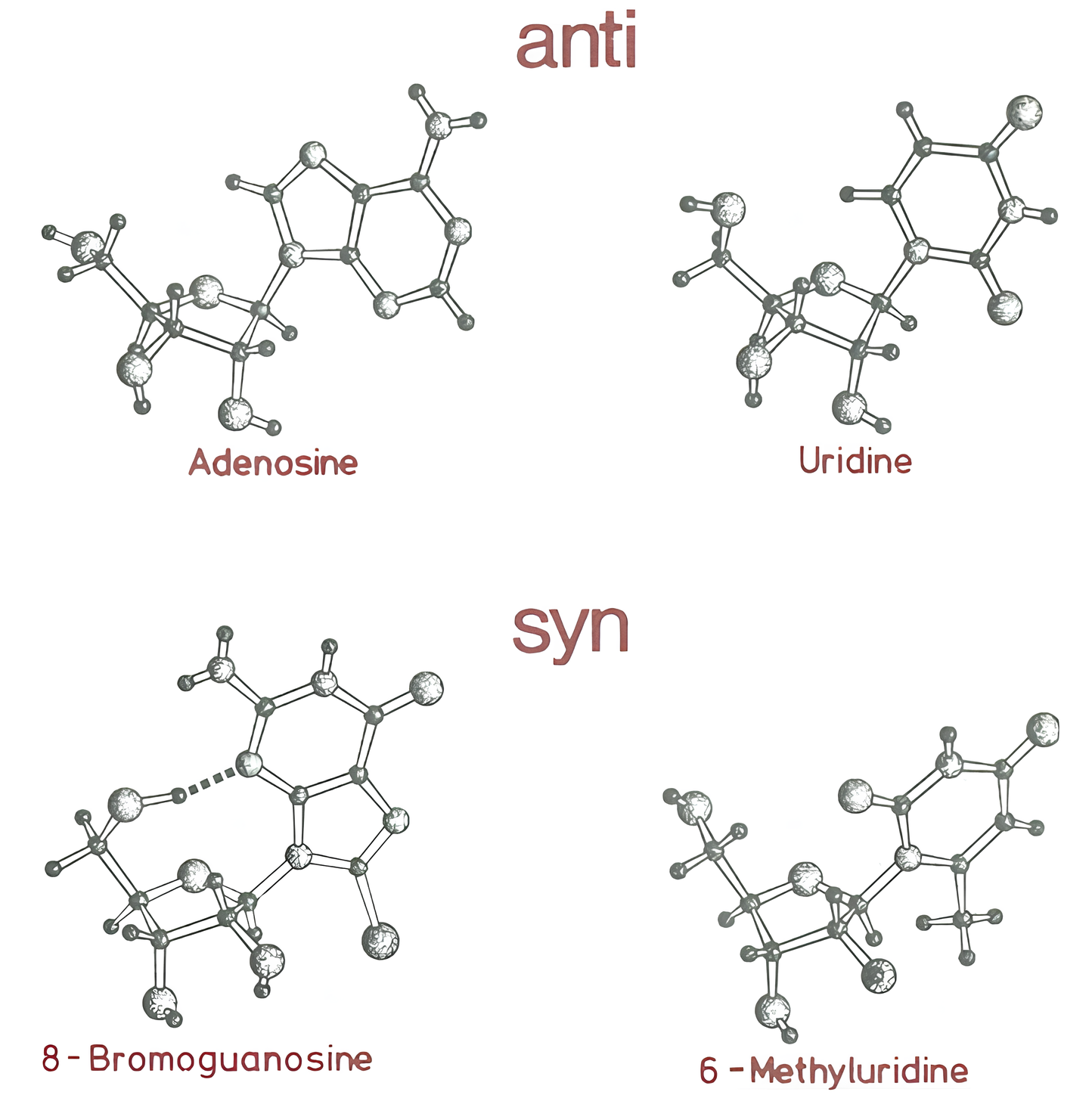
Presence of base triplets with syn glycosidic bond angle
Dihedral angles are stable along the simulation
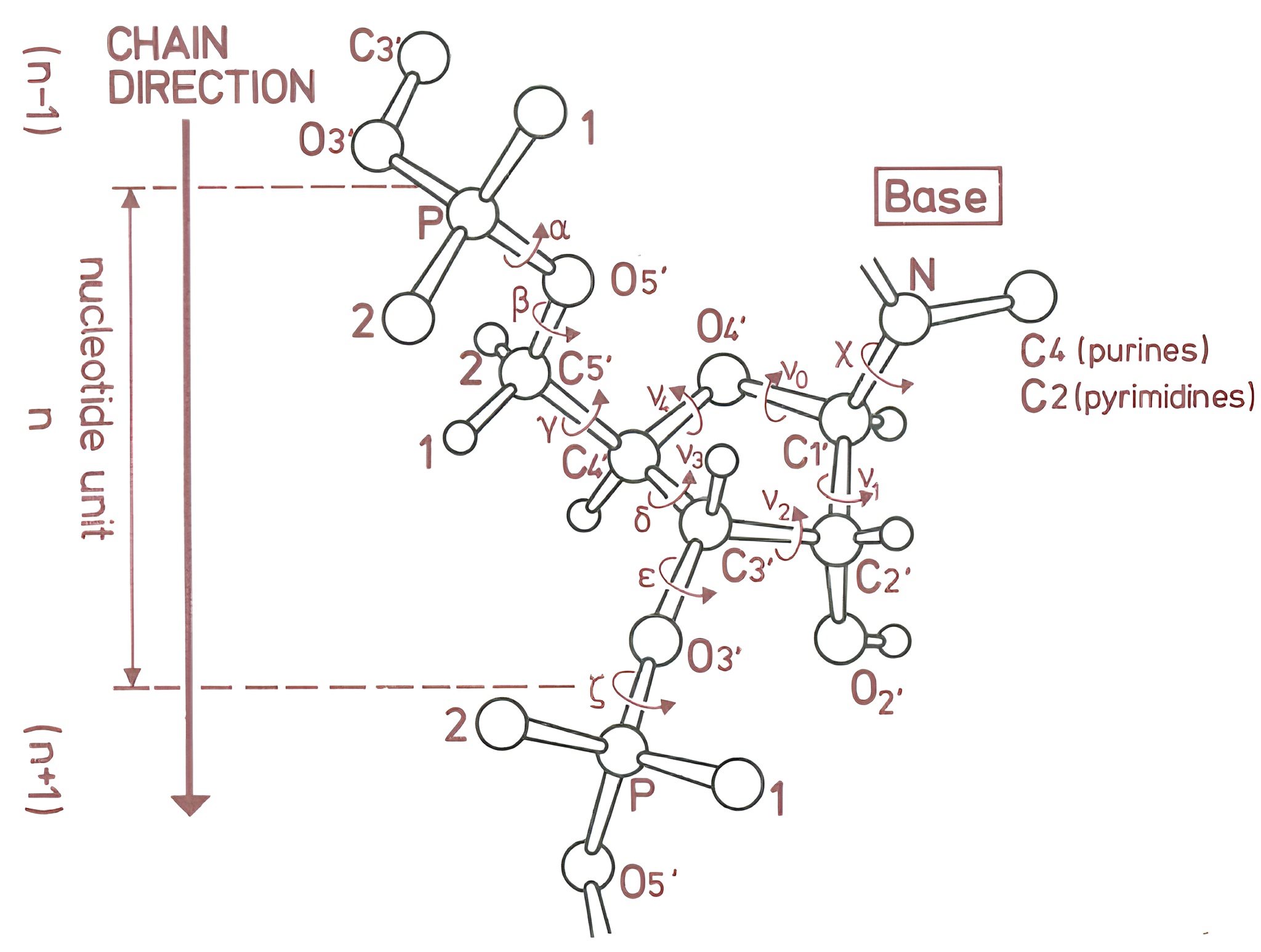


Binding requires non-canonical puckers
\[\color{#F0A308}\theta_M \color{#000000}= \frac{\color{#376bd3}\nu_2}{\cos \color{#279400}P}\]
\[\tan \color{#279400}P \color{#000000}= \frac{(\color{#008594}\nu_4 \color{#000000}+ \color{#A91FB6}\nu_1\color{#000000})-(\color{#940000}\nu_3 \color{#000000}+ \color{#947E00}\nu_0\color{#000000})}{2\color{#376bd3}\nu_2\color{#000000}(\sin(\frac{\pi}{5}) + \sin(\frac{2\pi}{5}))}\]
Binding requires non-canonical puckers
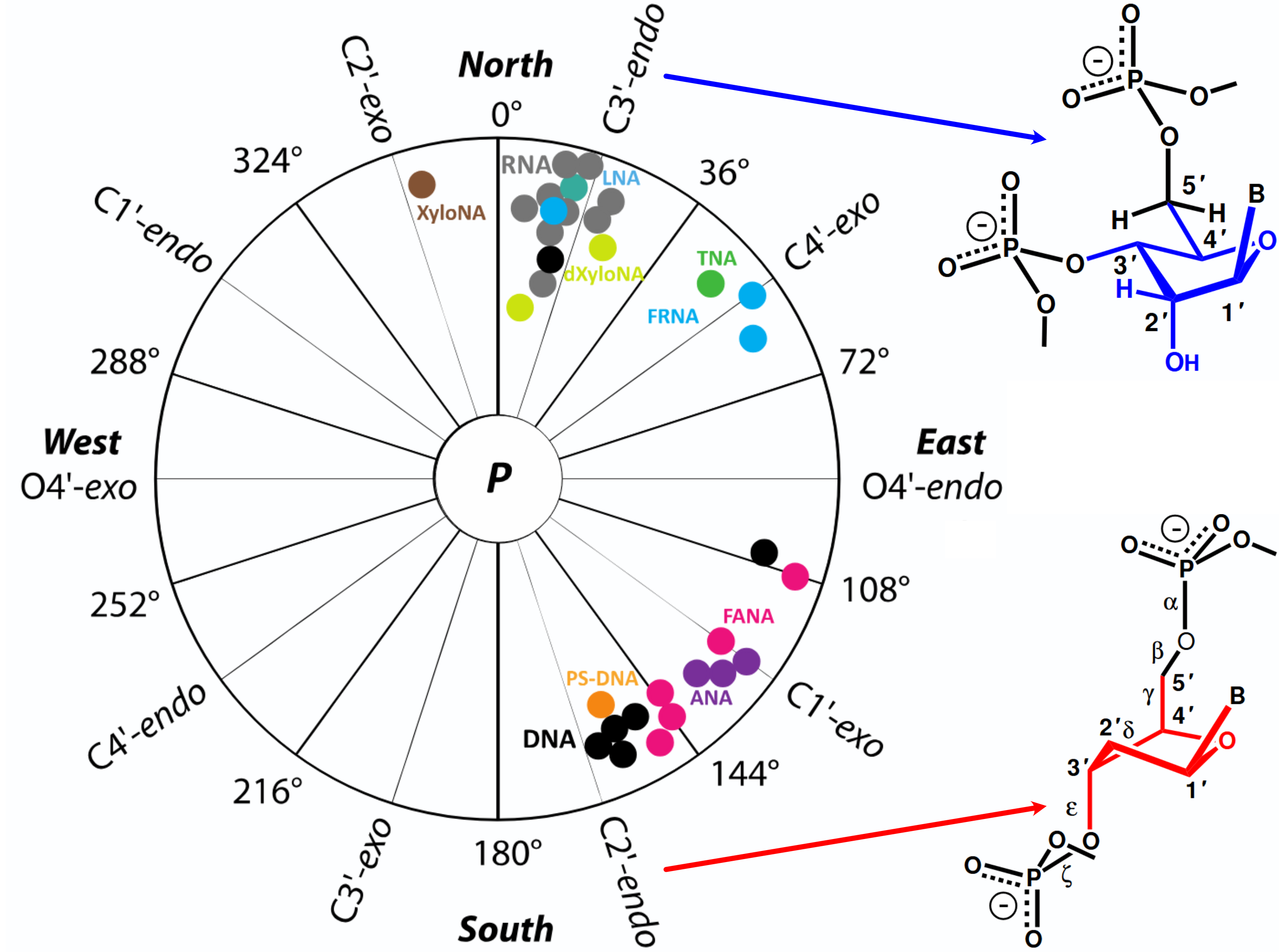
Binding requires non-canonical puckers
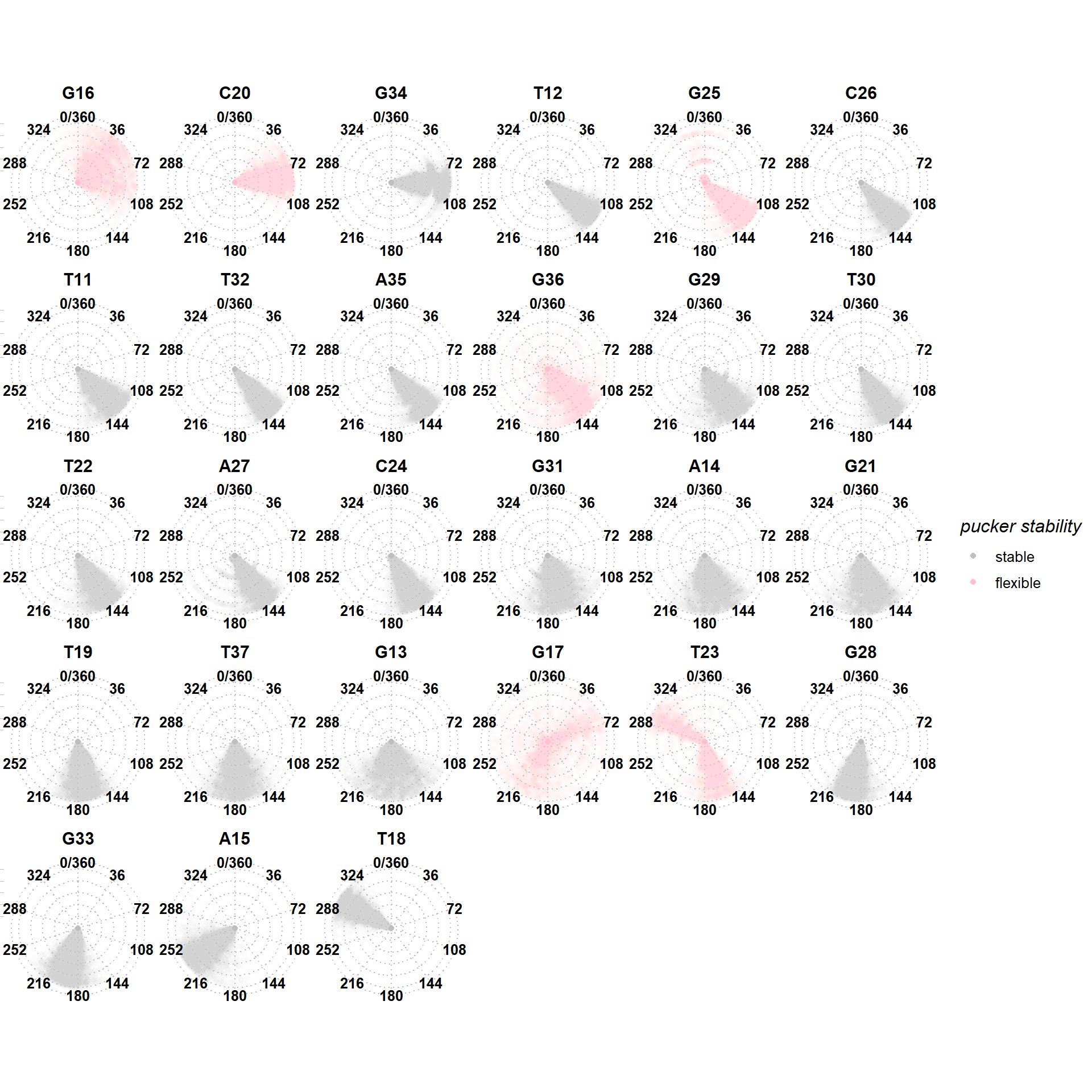
Binding requires non-canonical puckers

T18 adopts a C1’-endo pucker upon binding
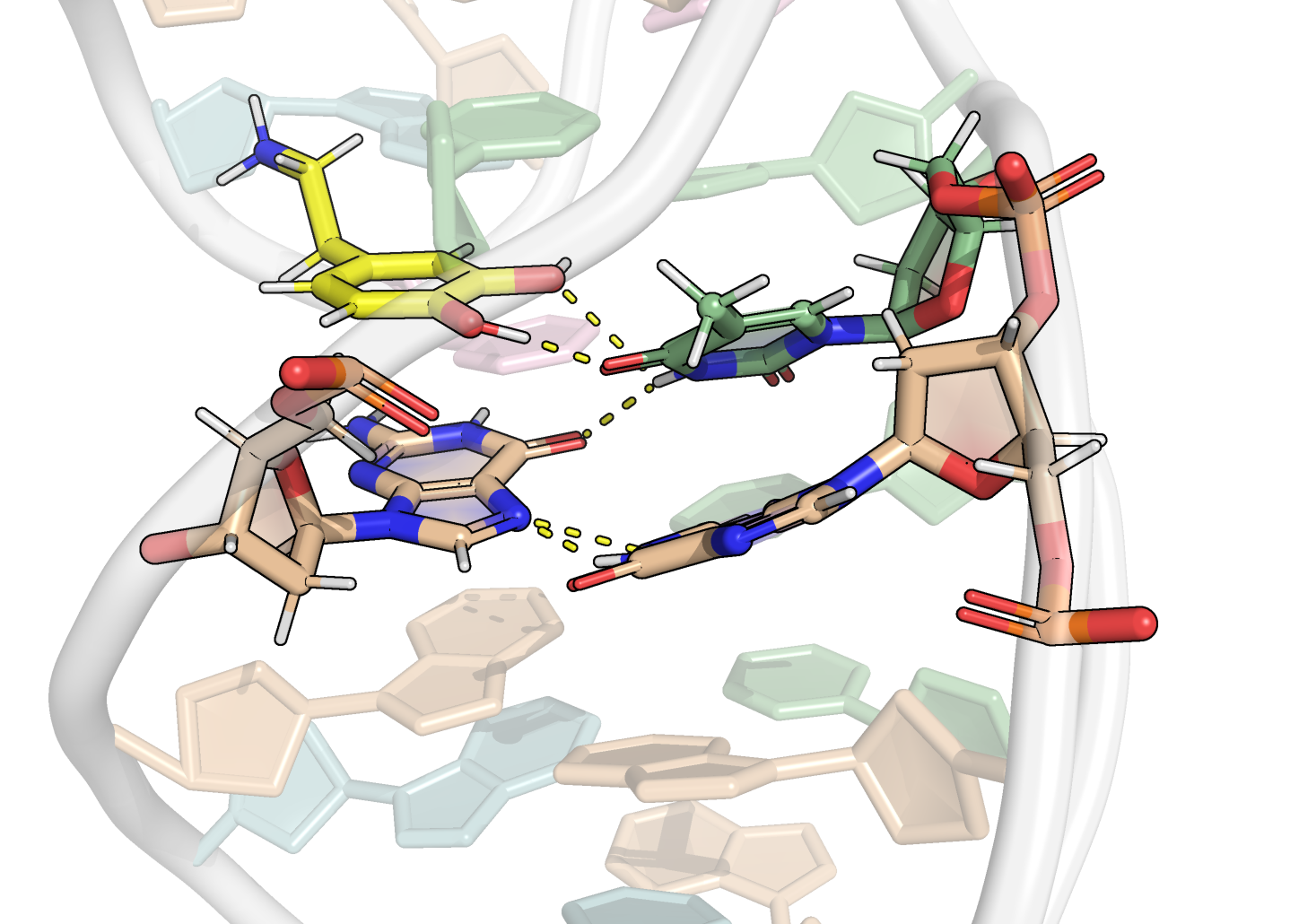
Binding through H-bonds, electrostatic and π interactions
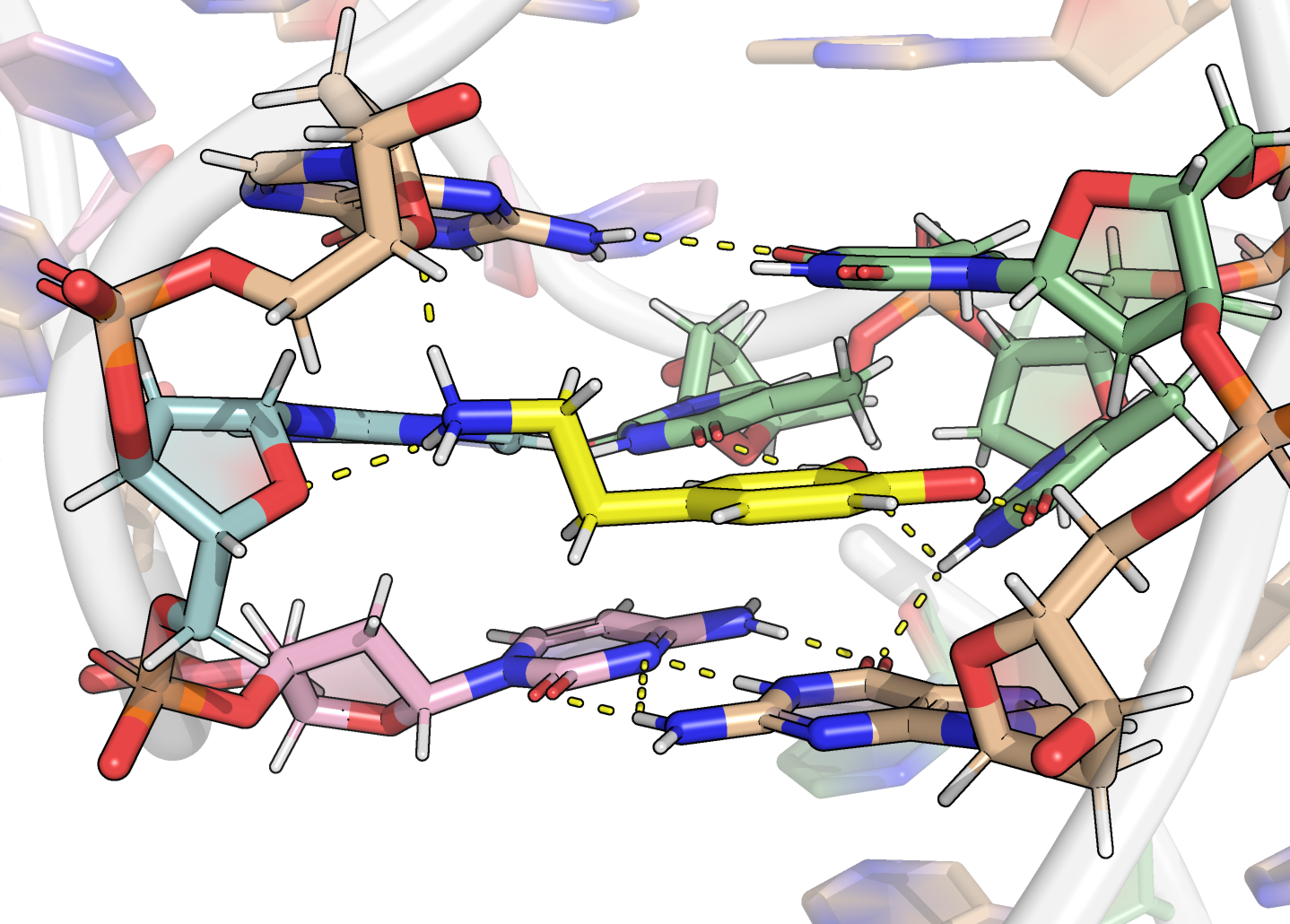
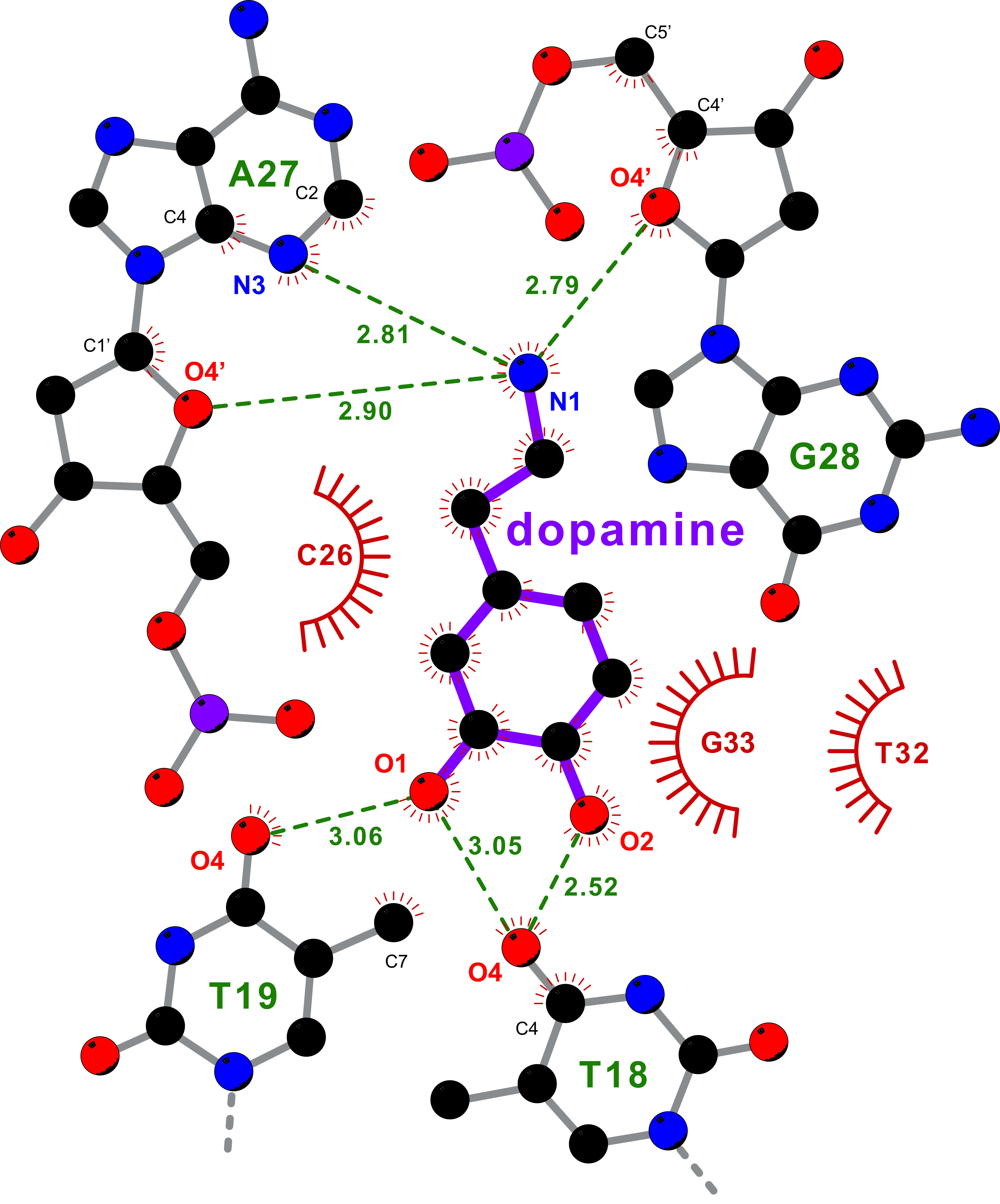

Restraints are necessary to refine the binding mode
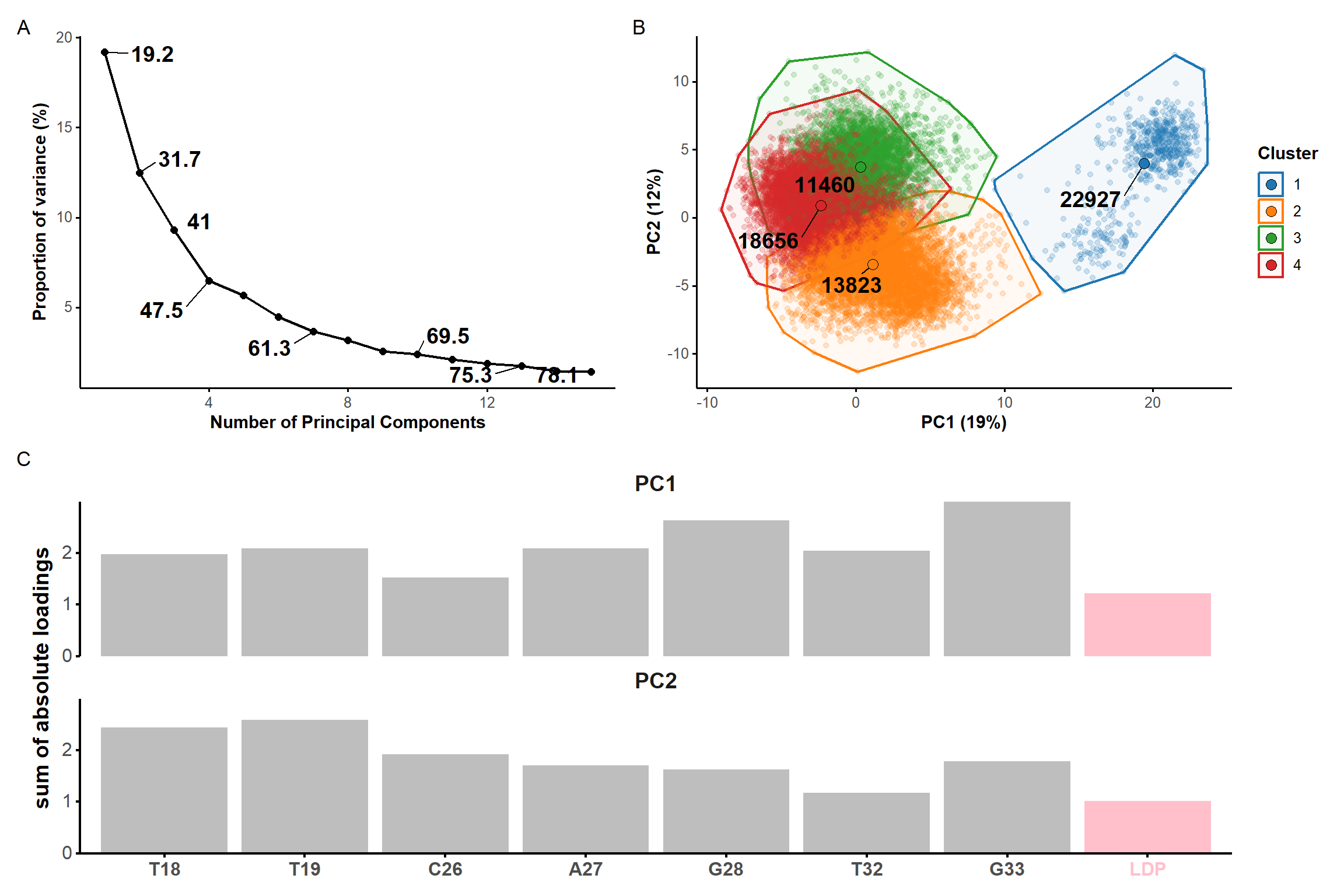
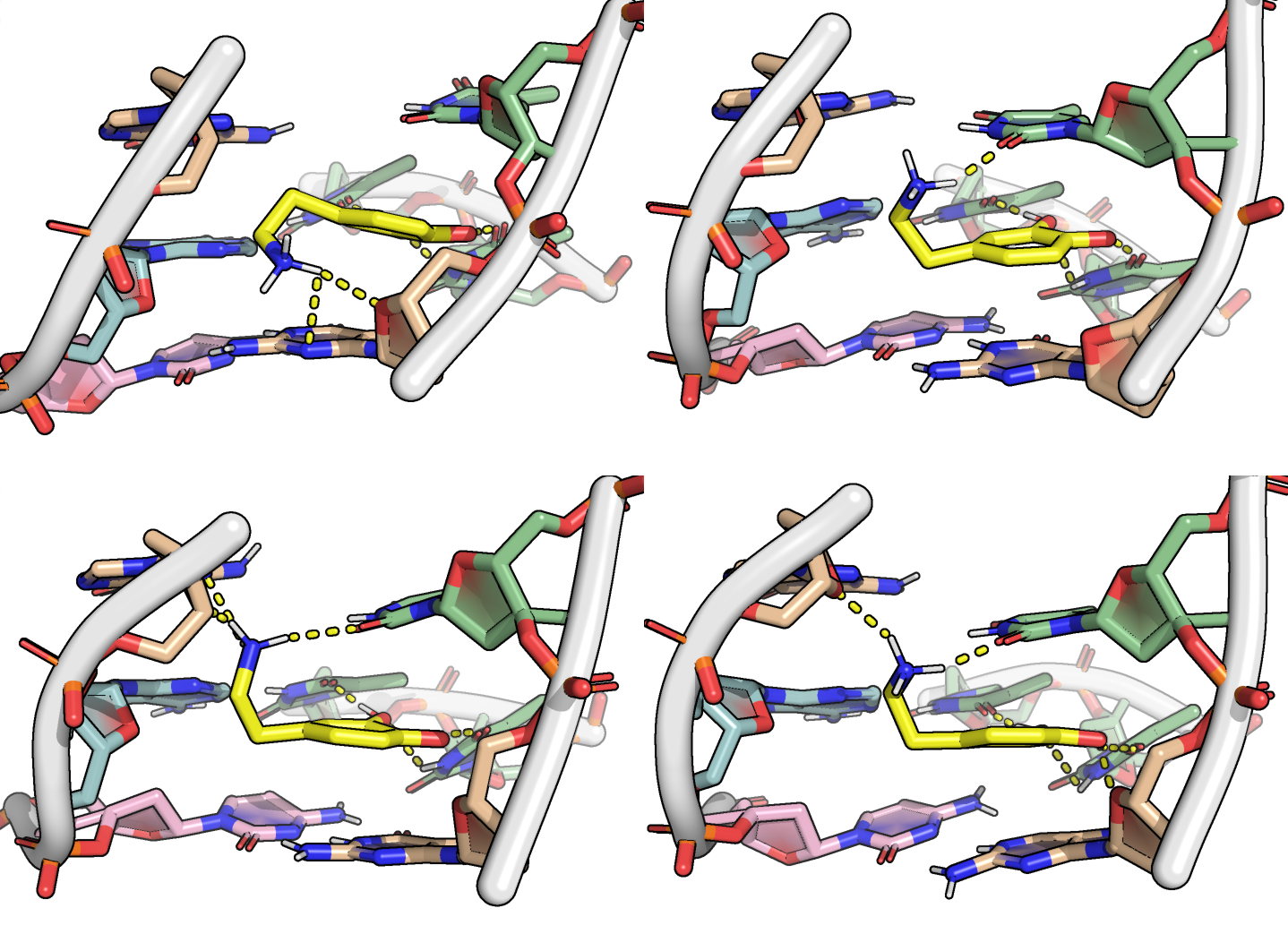
H-bonds inform structural dynamics
and model robustness
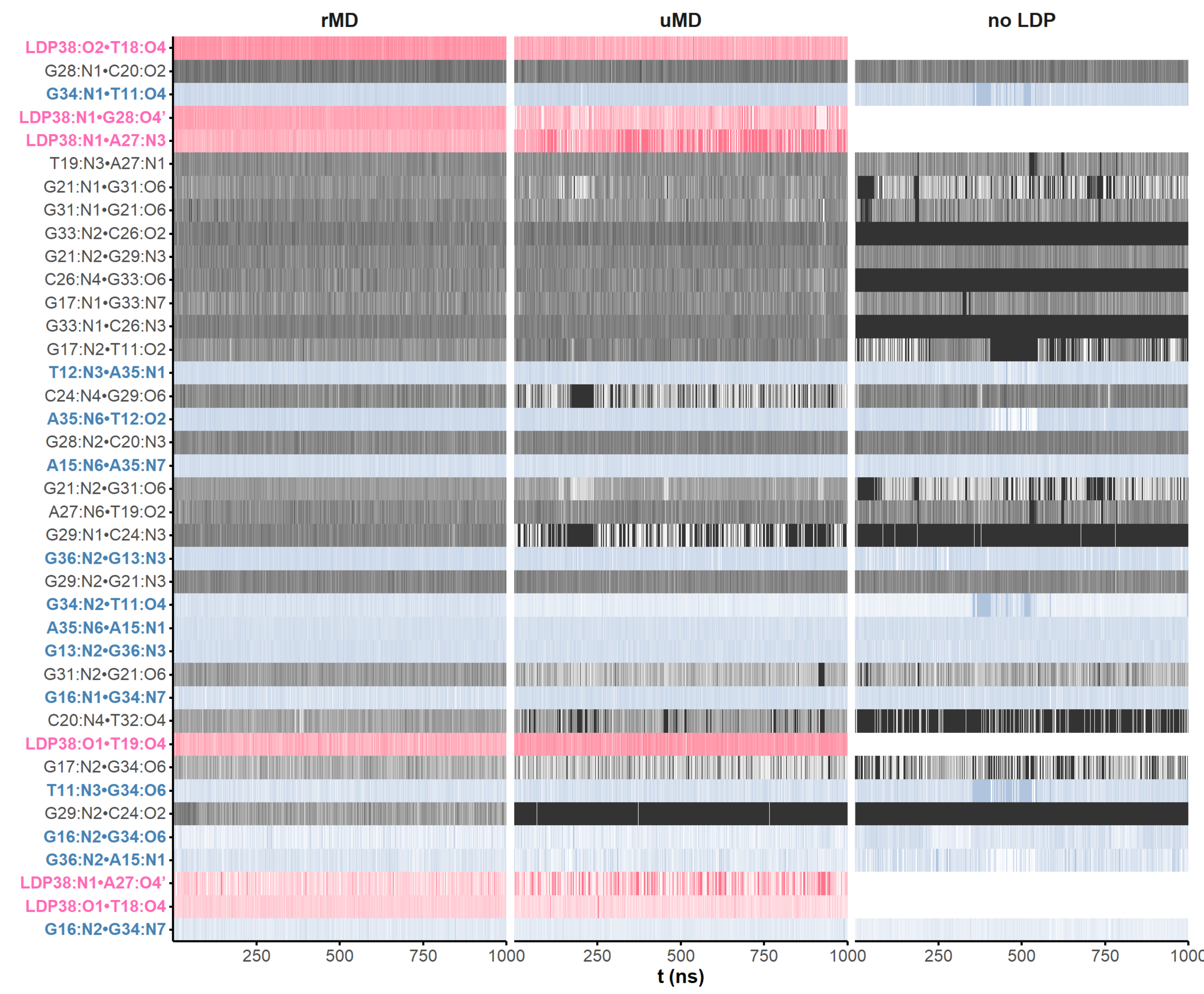
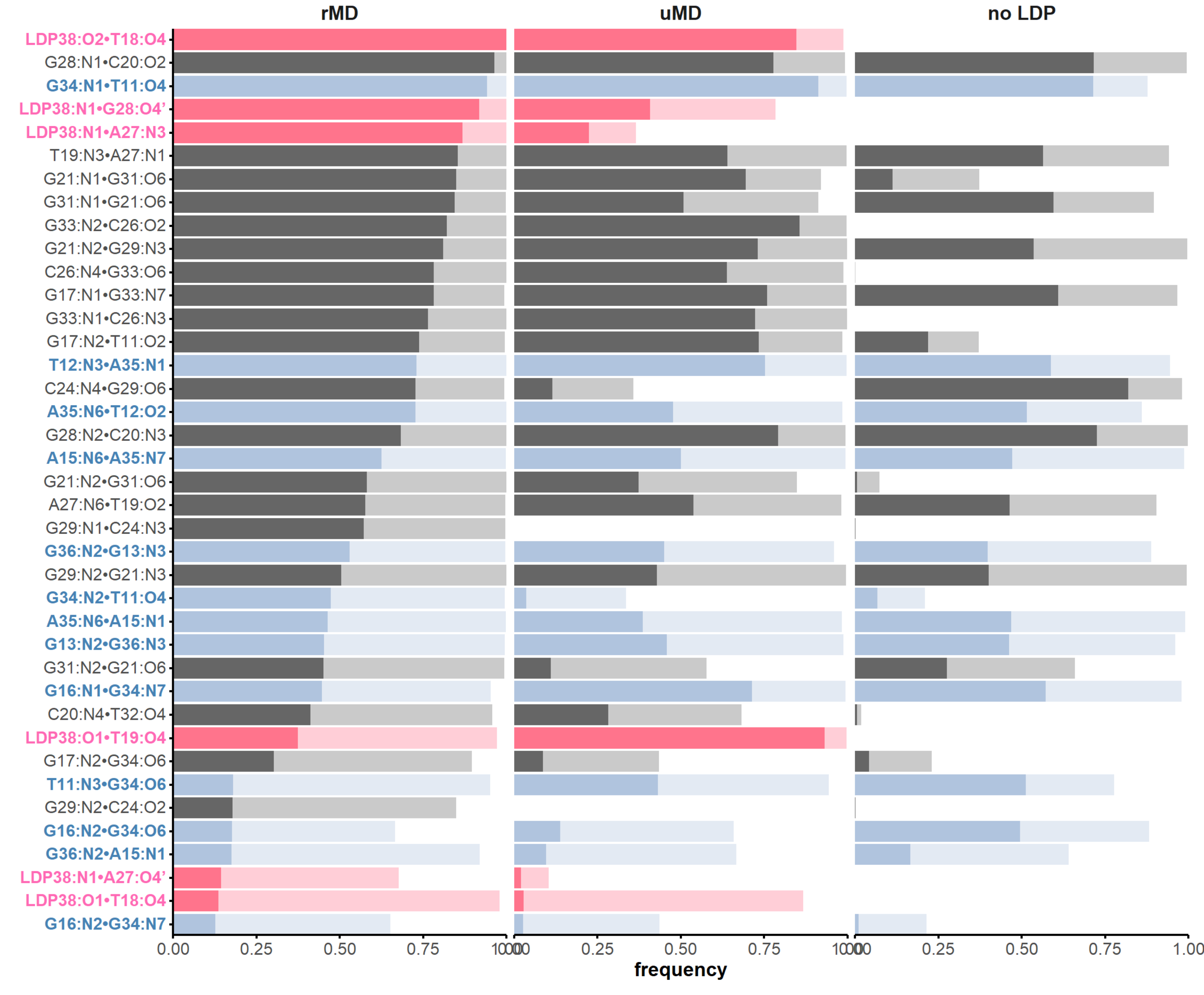
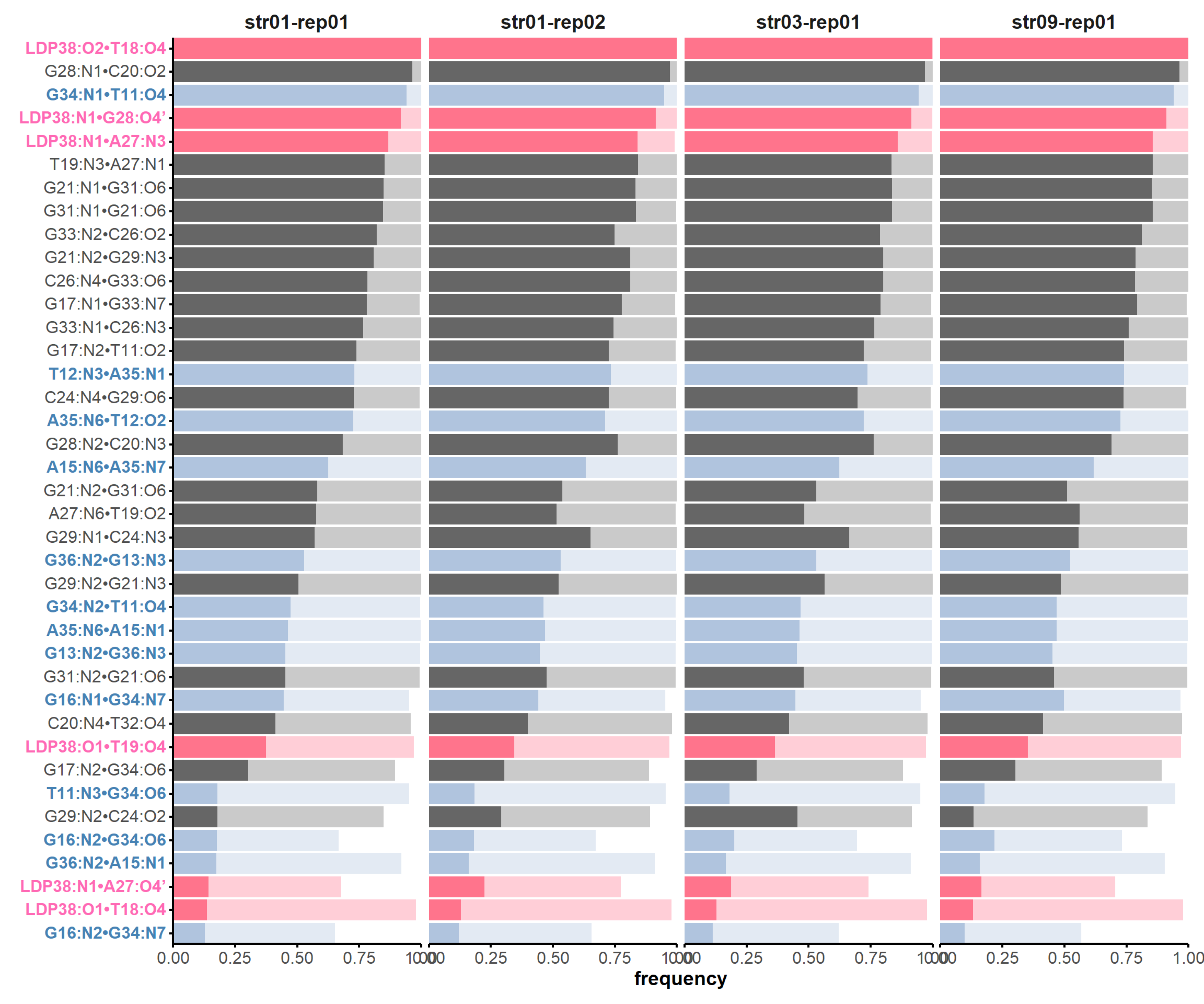
Prediction models still come short for aptamers

- 0 out of 107 submitted models with \(RMSD < 10 Å\)
- Training set is not diverse enough
- Dopamine essential for folding
